

RESEARCH
The HIMS-Biocat group is mainly working on four complementary and interconnected topics (Fig. 1). We work on fundamental studies of enzyme catalysis and mechanisms and this generates the required knowledge that we need for performing enzyme engineering. The enzyme engineering work can have several aims such as to create new enzymes whose activity is not known in nature; to expand the substrate scope and/or improve the activity of existing enzymes; to increase the enzyme stability to fulfil the need for a chemical process. Upon successful development, these engineered enzymes can also be combined with already available enzymes in our lab to generate multi-enzymatic cascades for the conversion of inexpensive and available starting material into high-value chemical products. We can run these biocatalytic cascade reactions both in batch and in continuous flow systems. For this purpose, we are working on projects aimed at facilitating enzyme immobilisation procedures along with improved stability and retention of catalytic activity. We have also recently been using our immobilisation techniques to create biocatalytic systems that find application as either electrochemical biosensors or to perform bio-electrochemical reactions. Finally, another topic is the development of multi-enzymatic cascades into living cells, thus creating artificial metabolic pathways in vivo.
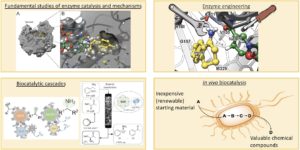
Figure 1
Brief account of selected research by HIMS-Biocat group during the past six years
1) Protein engineering of oxidoreductases
One of our target molecules are chiral amines. In fact, amines—particularly α-chiral amines—are widely used chemical intermediates for the production of active pharmaceutical ingredients, fine chemicals, and agrochemicals. In particular, α-chiral amines constitute approximately 40% of the optically active drugs that are currently primarily commercialised as single enantiomers. These molecules are classically synthesised in industry starting from prochiral ketones through multi-step chemical routes that generate copious amounts of waste; this production is also based on the use of increasingly unsustainable catalysts in the pivotal stereoselective steps. In contrast, amine dehydrogenases (AmDHs) are enzymes that catalyse the asymmetric reductive amination in aqueous buffer, under mild conditions and at the sole expense of ammonia and a hydride source (e.g., formate). However, the diversity of substrate scope and activity among AmDHs was (and still partly is) constrained. With the aim of addressing this limitation, we created a new family of AmDHs that can aminate pharmaceutically relevant aromatic ketones with high activity and stereoselectivity (Nature Commun. 2019, 10, 3717). The enzyme engineering project was guided rationally by structural information obtained from computational models. Interestingly, the AmDHs were created from an enzyme scaffold that does not catalyse any asymmetric transformation in nature. The best variant LE-AmDH-v1 displays a unique substrate-dependent switch of enantiospecificity (i.e., enantiomeric excess was >99% in most case, either R or S-selective), is highly thermostable (Tm of 69 °C), and retains almost entirely its catalytic activity upon incubation up to 50 °C for several days.
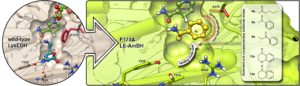
Figure 2
Stereoselective epoxidation of alkenes using molecular oxygen is another highly sought chemical transformation that we pursue in our lab. With the aim of improving the aerobic biocatalytic epoxidation, we created a chimeric styrene monooxygenase enzyme (Fus-SMO) that enables the stereoselective epoxidation of styrene derivatives in a whole cell system and at preparative scale by consuming only molecular oxygen and stoichiometric amounts of formate salt (Figure3; ChemBioChem 2018, 19, 679). The chimeric construct consists of a genetic fusion between two enzymatic units, namely the reductive and the epoxidating units, through a designed flexible linker. Thus, catalytic efficiency is enhanced because of a more balanced expression level between the two fused enzymatic units and the improved transfer of the reduced flavin cofactors between them during the catalytic cycle.

Figure 3
2) Asymmetric reductive amination of ketones
Amine dehydrogenases (AmDHs) catalyse the reductive amination of carbonyl compounds in aqueous buffer, under mild conditions and at the sole expense of ammonia and a hydride source. We have elucidated the substrate scope and the catalytic properties of synthetically useful and previously available AmDHs (Green Chem. 2017, 19, 453) and, as mentioned above, created a new family of AmDHs with improved catalytic properties and expanded substrate scope, particularly towards pharmaceutically relevant and aromatic ketones (Nature Commun. 2019, 10, 3717). In our optimised set-up, we carry out the reductive amination in ammonium formate buffer that acts simultaneously as reaction buffer, source of amine donor and hydride. In fact, the reducing equivalents are coming from the formate that is oxidized to bicarbonate by a formate dehydrogenase enzyme (Fig. 4).

Figure 4
The above described AmDHs perform the reductive amination of ketones to form (R)-configured amines in excellent enantiomeric excess. However, we could also access (S)-configured amines by performing the kinetic resolution of racemic amines (Fig. 5; ChemCatChem 2020, 12, 2184 ). This was possible by combining the AmDH with a NADH‐oxidase (NOx). Using these optimal conditions, the kinetic resolution of a number of substituted α‐methylbenzylamines, 1‐aminotetralin, 1‐aminoindane, and 4‐aminochromane was complete (i.e., according to optimal theoretical conversion of ca. 50%) with up to >99% enantiomeric excess.
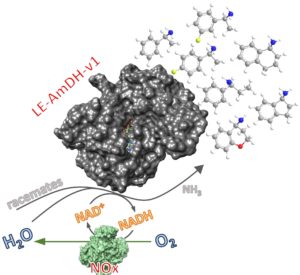
Figure 5
However, we also investigated other synthetically useful enzymes for α-chiral amine synthesis from ketones such ω-transaminases (ωTAs). These enzymes catalyse the stereoselective transfer of an amine group from an amine donor to a ketone acceptor via the pyridoxal-5’-phosphate cofactor. This natural reaction can be exploited either for the kinetic resolution of a racemic amine using pyruvate as sacrificial carbonyl-containing acceptor molecule, or for the formal asymmetric amination of a prochiral ketone using alanine or 2-propylamine as sacrificial amine donor. However, in aqueous environment and considering the most applied couple l- or d-alanine/pyruvate, the kinetic resolution is generally the more thermodynamically favoured process. ωTAs have been extensively applied in aqueous environment and in batch system both in academic and industrial settings. However, continuous flow biocatalysis has become increasingly popular because it combines the inherent selectivity of enzymes with practical flow systems to enable the gram-scale production of high-value chemicals. Continuous flow biocatalysis requires immobilisation of enzymes, thus enabling biocatalyst recyclability and often enhancing stability and increasing operational window. In this context, our group has studied the immobilisation of two stereocomplementary ωTAs onto polymer-coated controlled porosity glass beads (EziGTM) via FeIII-affinity-binding. Both immobilised ωTAs retained elevated activity when tested for the kinetic resolution of a racemic amine in multiple and consecutive batch experiments (Fig. 6; J. Biotechnol. 2019, 291, 52). The immobilised ωTAs was then applied in a continuous flow packed-bed reactor producing several grams of α-chiral amine in the kinetic resolution process (STY 335 g L-1 h-1).

Figure 6
Later, we presented the first account of applying immobilised ωTA enzymes in neat organic solvents and continuous flow system for the formal reductive amination of a prochiral ketone at the expense of stoichiometrically added 2-propylamine as sacrificial amine donor (Fig. 7, Adv. Synth. Catal. 2020, 362, 1858). Since dynamic and catalytic properties of enzymes in non-aqueous media are dependent on the presence of a limited but critical amount of water, we applied salt hydrate pairs during both the biocatalyst immobilisation step and the progress of the reaction. Optimal conditions were found with more hydrophobic carrier materials and toluene as reaction solvent. The applicability of the immobilised biocatalyst in neat organic solvents was further demonstrated in a continuous flow packed-bed reactor. This showed excellent performance without observable loss of enzymatic activity over several days of operation.
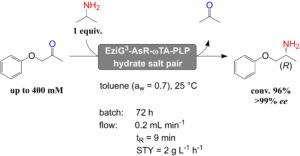
Figure 7
3) Biocatalytic synthesis of α-chiral amines from alcohols
In the context of multi-enzymatic and selective synthesis of α-chiral amines, our group has developed a biocatalytic cascade and related enzymes for the asymmetric conversion of alcohols into optically active amines (Science, 2015, 359, 1525). The method is based on the use of two enzymes, namely an alcohol dehydrogenase (ADH) and an amine dehydrogenase (AmDH) that operate in tandem with a perfect internal recycling of redox equivalents; thus, the process is entirely redox self-sufficient, also defined as a ‘hydrogen-borrowing’ process (or more properly ‘hydride-borrowing’ process). Notably, the biocatalytic method possesses the highest possible atom efficiency, sourcing nitrogen from ammonia and generating water as the sole by-product. Although this asymmetric biocatalytic hydrogen-borrowing amination of alcohols (including racemic alcohols) was initially conceived to be highly chemo- and stereo-selective, we have late introduced the capability of distinguishing between different hydroxyl functions within the same molecule (i.e., regioselectivity) as depicted in Figure 8A (Green Chem. 2019, 21, 6246-6251). The versatility of the biocatalytic alcohol amination process was also demonstrated in a work in which ADH-AmDH genes were introduced into E. coli to carry out this transformation in vivo (Figure 1B, Green Chem. 2019, 21, 3846). Using optimised genetic constructs for balanced expression levels of the dehydrogenases, the influence of several parameters towards the productivity of the system were investigated: the intracellular NAD+/NADH redox balance, the cell loading, the survival rate of cells, the possible toxicity of the components of the reaction, etc. The optimised E. coli/ADH-AmDH strains produced enantiopure amines from the alcohols with up to 80% conversion and a molar productivity up to 15 mM. Practical applicability was demonstrated in a gram-scale biotransformation.

Figure 8
In another type of synthetic approach, both dehydrogenases (ADH and AmDH) were selectively co-immobilised via FeIII-affinity-binding on controlled porosity glass carrier and applied for the ‘hydrogen-borrowing’ alcohol amination (Fig. 9; ChemCatChem 2018, 10, 731). The carrier material used possesses high porosity that enables a high enzyme loading capacity (typically 40% w/w), whereas the polymeric coating enhances the enzyme stability during biotransformations. Furthermore, the enzyme can be immobilised directly from the cell lysate hence not requiring any previous purification step. The performance of the heterogeneous immobilised dual-enzyme system was optimised in terms of catalysts loading, molar ratio between ADH and AmDH, total amount of enzymes per mass of affinity beads as well as substrate concentration. The recyclability of the dual-enzyme system was demonstrated leading to an up to 15-fold increased of chemical total turnover numbers (TTNs) compared with the reactions using free enzymes in solution.
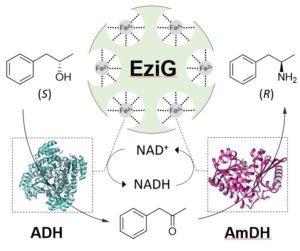
Figure 9
Always in the area of multi-enzymatic conversion of alcohols to amines, we have developed an orthogonal biocatalytic network for the quantitative and stereospecific amination of alcohols (Fig. 10; Org. Biomol. Chem. 2017, 15, 8313). The biocatalytic network consists of two modules. The first module, driven by a NADP-dependent alcohol dehydrogenase and a NADP-oxidase, oxidises racemic secondary or primary alcohols to ketones or aldehydes at the expense of molecular oxygen. The second module, driven by a NAD-dependent amine dehydrogenase and a formate dehydrogenase, performs the asymmetric amination of the carbonyl compound intermediate. As the two enzymatic modules have a divergent co-enzyme specificity (NAD vs NADP), multiple redox reactions run simultaneously in the same vessel without the need for a physical separation. This property in chemistry is defined as ‘orthogonality’. Orthogonal chemical reactions are very rare as traditional chemical (redox) reagents undergo cross-reactivity. In contrast, our system profits of the exquisite selectivity of enzymes to use and recycle a particular source of ‘nature hydride’ (NADH or NADPH). Thus, amines were obtained in >99% enantiomeric excess as a result of the perfect stereoselectivity of amine dehydrogenases.
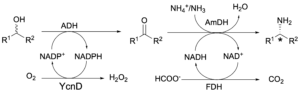
Figure 10
4) Studies on enzyme promiscuity and discovery of new biocatalytic reactions
The concept of enzyme promiscuity refers to an enzyme’s ability to catalyse mechanistically distinct reactions (i.e., catalytic promiscuity) or convert structurally diverse substrates by following the same mechanism (i.e., substrate promiscuity). The promiscuous catalytic behavior of enzymes has been harnessed for chemical synthesis and has served for the evolution of variants possessing novel catalytic activities and/or enhanced substrate scope. In some cases, the enzyme’s catalytic or substrate promiscuity can be simply tuned by changing the reaction conditions (i.e., condition promiscuity). During our studies on enzyme catalysis and mechanisms, we discover the promiscuous catalytic activity of an alcohol oxidase, namely a galactose oxidase, that catalyses the one-pot transformation of alcohols into nitriles under mild, cyanide-free conditions (Fig. 11; Angew. Chem. Int. Ed. 2018, 57, 14240). The advantage of the use of enzymes lies in the absence of toxic compounds (notably cyanide) and the use of an aqueous environment. Furthermore, the conversion proceeds under atmospheric pressures and requires less energy consumption (for heat and pressure) compared with chemical methods. Additionally, no chemical method for the conversion of alcohols into nitriles has a comparable atom efficiency, which means that the biocatalytic method generates less waste. Increasing the conversion above 99% and maximising chemoselectivity would lead to a reaction mixture of pure nitrile and eliminate the need for further purification steps, which we have already achieved with selected substrates.
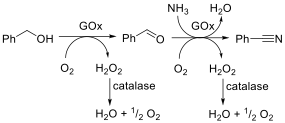
Figure 11
During our studies on amine dehydrogenases (AmDHs) for the synthesis of secondary and tertiary amines, we gained new insights into the catalytic mechanism of these enzymes (ChemBioChem, 2019, 20, 800). Results showed that AmDHs accept ammonia as well as more complex amines to enable the synthesis of secondary and tertiary amines in enantioenriched form. However, this transformation was often accompanied by a side reaction that led to the formation of the structurally related primary amines although ammonia was not present or generated in the reaction mixture. We concluded that this promiscuous activity is the result of the catalytic activity of the AmDHs used. In particular, the mechanism entails the formation of the imine intermediate in the active site of the enzymes, followed by an unprecedented cofactor (NAD) dependent isomerisation step. Then, the isomeric imine intermediate can undergo to hydrolysis due to the particular catalytic environment in the active site of the enzyme, thereby leading to the formation of the corresponding primary amine. In summary, with the aid of computational studies, we were able to rationalise the formation of all products observed in the reaction mixture as well as to understand the chemo- and enantio-selective preferences of the AmDHs. The study concluded with a proposed overall reaction mechanism as depicted in Figure 12.

Figure 12
As mentioned in section 1, we have engineered a l-lysine-(ε-deaminating) dehydrogenase (LysEDH) from Geobacillus s. to create amine dehydrogenase (AmDH) variants that convert aromatic ketones into α-chiral amines with excellent enantioselectivity. Additionally, we revealed the catalytic promiscuity of the LysEDH since this dehydrogenase was also capable of reducing aromatic aldehydes into primary alcohols. Therefore, we harnessed the promiscuous alcohol dehydrogenase (ADH) activity of LysEDH to create new variants that exhibited enhanced catalytic activity for the reduction of substituted benzaldehydes and arylaliphatic aldehydes into primary alcohols (Chem Eur. J. 2021). Notably, these novel engineered dehydrogenases also catalysed the reductive amination of a variety of aldehydes and ketones with excellent enantioselectivity, thus exhibiting a dual AmDH/ADH activity (Fig. 13). We applied the catalytic bi-functionality of these enzymes to perform an unprecedented one-pot ‘hydrogen-borrowing’ cascade to convert benzyl alcohol to benzylamine using a single enzyme. In summary, this work provided the first examples of enzymes showing ‘alcohol aminase’ activity.
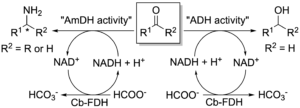
Figure 13
5) Chemoselective oxidation of aldehyde moieties to carboxylic acids
The chemoselective oxidation of aldehydes to carboxylic acids is an industrially relevant chemical reaction, particularly with respect to the synthesis of pharmaceuticals and bio-based polymers. Current oxidation procedures require the abundant use of toxic chemical reagents and often produce unwanted side-products. As an economically viable procedure that is based on environmentally benign reagents and/or solvents and combines an elevated productivity with a perfect selectivity was not available, our group developed a chemoselective bio-oxidation of aldehydes to carboxylic acids based on the use of aldehyde dehydrogenases (ALDHs) and nicotinamide adenine dinucleotide oxidase (NOx) either as isolated enzymes or in a whole cell system. The biocatalytic reaction operates in an aqueous buffer at 40 °C under atmospheric pressure and requires only air as the oxidant (Fig. 14; Green Chem. 2018, 20, 3931). To investigate the synthetic potential of the recombinant ALDHs, we performed an extensive study on sixty-one structurally diverse aldehydes. The majority of these substrates (aliphatic, aryl aliphatic, benzylic-, hetero-aromatic and bicyclic aldehydes) were converted with yields of well over 60% and in many cases even over 99%. In all cases, the chemoselectivity was perfect: no other product was detected except the expected carboxylic acid. This means that other oxidisable functionalities (such as the hydroxyl moiety, alkene groups, aryl groups, and sulphur as well as nitrogen heteroatoms) remained untouched. In particular, bio-based 5-(hydroxymethyl)furfural was converted into 5-(hydroxymethyl)furoic acid, in a two gram-scale reaction with perfect chemoselectivity and 61% isolated yield. 5-(hydroxymethyl)furoic acid finds application as building block for the production of bio-based polymers and pharmaceuticals.
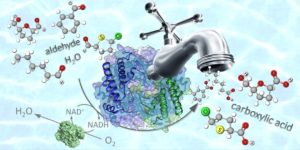
Figure 14