


PUBLICATIONS
2022
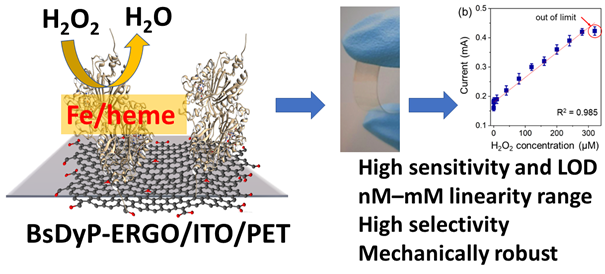
*Bacterial Peroxidase on Electrochemically Reduced Graphene Oxide for Highly Sensitive H2O2 Detection*
Bhardwaj, S. K.; Knaus, T.; Garcia, A.; Yan, N.; Mutti, F. G.*; ChemBioChem, 2022, 23, e202200346. DOI: 10.1002/cbic.202200346
morePeroxidase enzymes enable the construction of electrochemical sensors for highly sensitive and selective quantitative detection of various molecules, pathogens and diseases. Herein, we describe the immobilization of a peroxidase from Bacillus s. (BsDyP) on electrochemically reduced graphene oxide (ERGO) deposited on indium tin oxide (ITO) and polyethylene terephthalate (PET) layers. XRD, SEM, AFM, FT-IR and Raman characterization of the sensor confirmed its structural integrity and a higher enzyme surface occupancy. The BsDyP-ERGO/ITO/PET electrode performed better than other horseradish peroxidase-based electrodes, as evinced by an improved electrochemical response in the nanomolar range (linearity 0.05–280 μM of H2O2, LOD 32 nM). The bioelectrode was mechanically robust, active in the 3.5–6 pH range and exhibited no loss of activity upon storage for 8 weeks at 4 °C.
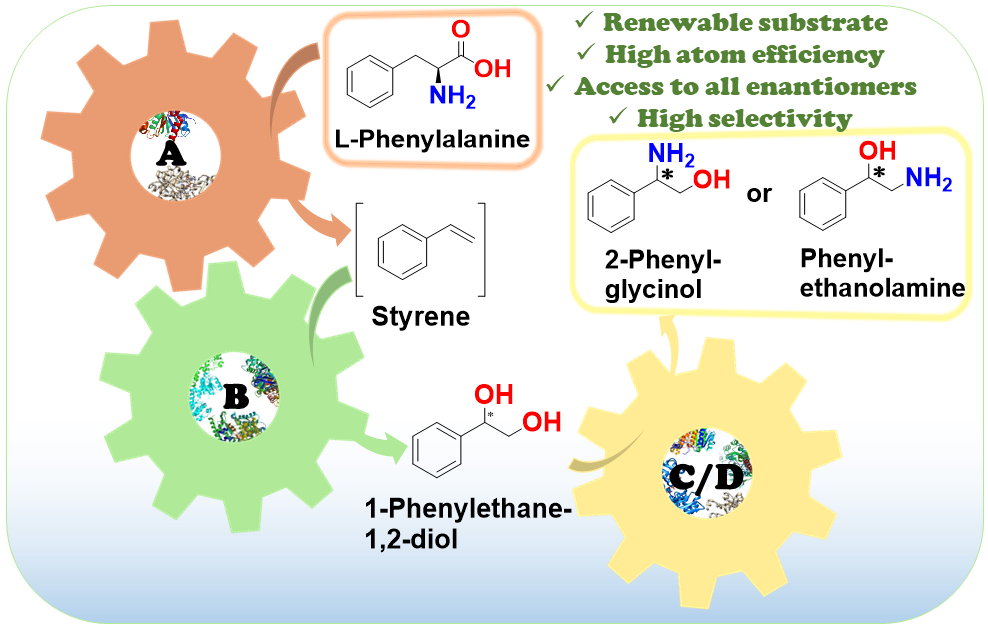
*High-Yield Synthesis of Enantiopure 1,2-Amino Alcohols from l-Phenylalanine via Linear and Divergent Enzymatic Cascades*
Corrado, M. L.; Knaus, T.; Schwaneberg, U.; Mutti, F. G.*; Org. Process Res. Dev., 2022, 26, 2085-2095. DOI: 10.1021/acs.oprd.1c00490
moreEnantiomerically pure 1,2-amino alcohols are important compounds due to their biological activities and wide applications in chemical synthesis. In this work, we present two multienzyme pathways for the conversion of l-phenylalanine into either 2-phenylglycinol or phenylethanolamine in the enantiomerically pure form. Both pathways start with the two-pot sequential four-step conversion of l-phenylalanine into styrene via subsequent deamination, decarboxylation, enantioselective epoxidation, and enantioselective hydrolysis. For instance, after optimization, the multienzyme process could convert 507 mg of l-phenylalanine into (R)-1-phenyl-1,2-diol in an overall isolated yield of 75% and >99% ee. The opposite enantiomer, (S)-1-phenyl-1,2-diol, was also obtained in a 70% yield and 98–99% ee following the same approach. At this stage, two divergent routes were developed to convert the chiral diols into either 2-phenylglycinol or phenylethanolamine. The former route consisted of a one-pot concurrent interconnected two-step cascade in which the diol intermediate was oxidized to 2-hydroxy-acetophenone by an alcohol dehydrogenase and then aminated by a transaminase to give enantiomerically pure 2-phenylglycinol. Notably, the addition of an alanine dehydrogenase enabled the connection of the two steps and made the overall process redox-self-sufficient. Thus, (S)-phenylglycinol was isolated in an 81% yield and >99.4% ee starting from ca. 100 mg of the diol intermediate. The second route consisted of a one-pot concurrent two-step cascade in which the oxidative and reductive steps were not interconnected. In this case, the diol intermediate was oxidized to either (S)- or (R)-2-hydroxy-2-phenylacetaldehyde by an alcohol oxidase and then aminated by an amine dehydrogenase to give the enantiomerically pure phenylethanolamine. The addition of a formate dehydrogenase and sodium formate was required to provide the reducing equivalents for the reductive amination step. Thus, (R)-phenylethanolamine was isolated in a 92% yield and >99.9% ee starting from ca. 100 mg of the diol intermediate. In summary, l-phenylalanine was converted into enantiomerically pure 2-phenylglycinol and phenylethanolamine in overall yields of 61% and 69%, respectively. This work exemplifies how linear and divergent enzyme cascades can enable the synthesis of high-value chiral molecules such as amino alcohols from a renewable material such as l-phenylalanine with high atom economy and improved sustainability.
2021
*Enzymes Applied to the Synthesis of Amines*
Mutti, F. G.; Knaus T.; Chapter 6, in Biocatalysis for Practitioners. Techniques, Reactions and Applications, (Ed. de Gonzalo, G.; Lavandera, I.), Wiley, Weinheim (Germany), 2021, pages 143–180. ISBN: 978-3-527-34683-7.
moreA great variety of biocatalytic methods are available for the synthesis of amines, in particular α-chiral amines. Therefore, chapter 6 aims to orient non-specialists to the available biocatalytic methodologies for the synthesis of α-chiral amines with a focus on the enzymes’ substrate scope and selectivity as well as the structural diversity of the obtained products. The presented enzymatic methods comprise the use of hydrolases, amine oxidases, transaminases, amine dehydrogenases, imine reductases, ammonia lyases, Pictet-Spenglerases and engineered cytochromes P450. Classification and reactivity, biocatalytic applications and selected important examples are discussed for each enzyme category. Methodologies entail the kinetic resolution or deracemisation of racemic amines and the asymmetric synthesis from prochiral precursors, where applicable. Finally, the last chapter’s section illustrates practical procedures. Notably, the presented biocatalytic methods can yield primary, secondary and tertiary amine products and often with excellent chemo-, regio- and stereoselectivity under mild reaction conditions.
*High Regio- and Stereoselective Multi-enzymatic Synthesis of All Phenylpropanolamine Stereoisomers from β-Methylstyrene*
Corrado, M. L.; Knaus, T.; Mutti, F. G.*; ChemBioChem, 2021, 22, 2345–2350. DOI: 10.1002/cbic.202100123
moreWe present a one-pot cascade for the synthesis of phenylpropanolamines (PPAs) in high optical purities (er and dr up to >99.5 %) and analytical yields (up to 95 %) by using 1-phenylpropane-1,2-diols as key intermediates. This bioamination entails the combination of an alcohol dehydrogenase (ADH), an ω-transaminase (ωTA) and an alanine dehydrogenase to create a redox-neutral network, which harnesses the exquisite and complementary regio- and stereo-selectivities of the selected ADHs and ωTAs. The requisite 1-phenylpropane-1,2-diol intermediates were obtained from trans- or cis-β-methylstyrene by combining a styrene monooxygenase with epoxide hydrolases. Furthermore, in selected cases, the envisioned cascade enabled to obtain the structural isomer (1S,2R)-1-amino-1-phenylpropan-2-ol in high optical purity (er and dr >99.5 %). This is the first report on an enzymatic method that enables to obtain all of the four possible PPA stereoisomers in great enantio- and diastereo-selectivity.
*Generation of oxidoreductases with dual alcohol dehydrogenase and amine dehydrogenase activity*
Tseliou, V.; Schilder, D.; Masman, M. F.; Knaus, T.; Mutti, F. G.* Chem. Eur. J., 2021,27, 3315-3325. DOI: 10.1002/chem.202003140 (HOT PAPER)
moreThe l‐lysine‐ϵ‐dehydrogenase (LysEDH) from Geobacillus stearothermophilus naturally catalyzes the oxidative deamination of the ϵ‐amino group of l‐lysine. We previously engineered this enzyme to create amine dehydrogenase (AmDH) variants that possess a new hydrophobic cavity in their active site such that aromatic ketones can bind and be converted into α‐chiral amines with excellent enantioselectivity. We also recently observed that LysEDH was capable of reducing aromatic aldehydes into primary alcohols. Herein, we harnessed the promiscuous alcohol dehydrogenase (ADH) activity of LysEDH to create new variants that exhibited enhanced catalytic activity for the reduction of substituted benzaldehydes and arylaliphatic aldehydes to primary alcohols. Notably, these novel engineered dehydrogenases also catalyzed the reductive amination of a variety of aldehydes and ketones with excellent enantioselectivity, thus exhibiting a dual AmDH/ADH activity. We envisioned that the catalytic bi‐functionality of these enzymes could be applied for the direct conversion of alcohols into amines. As a proof‐of‐principle, we performed an unprecedented one‐pot “hydrogen‐borrowing” cascade to convert benzyl alcohol to benzylamine using a single enzyme. Conducting the same biocatalytic cascade in the presence of cofactor recycling enzymes (i.e., NADH‐oxidase and formate dehydrogenase) increased the reaction yields. In summary, this work provides the first examples of enzymes showing “alcohol aminase” activity.
2020
*Aerobic synthesis of aromatic nitriles from alcohols and ammonia using galactose oxidase*
Vilím, J.; Knaus, T.; Mutti, F. G.; Chapter 11.7 in Applied Biocatalysis: The Chemist's Enzyme Toolkit, (Eds. Whittall, J.; Sutton P.W.), John Wiley & Sons, Inc, 2020, pages 449-455. ISBN: 978-1-119-48701-2
moreThe enzymatic synthesis of aromatic nitriles from alcohols and ammonia presents a valuable alternative to currently applied chemical methodologies. The synthesis of nitriles is traditionally accomplished through non-environmentally friendly approaches such as transition metal catalyzed cyanation, Sandmeyer reaction or Rosenmund-von Braun reaction. Biocatalytic routes towards the synthesis of nitriles utilize either cyanide in combination with aldehydes (e.g., hydroxynitrile lyases, halohydrin dehalogenases) or require previous in-situ formation of the substrate (aldoxime dehydratases). Ammoxidation is a more sustainable alternative, as it consumes molecular oxygen and ammonia as reagents.
Galactose oxidase (GOx) is a copper-dependent oxidoreductase that naturally catalyzes the aerobic oxidation of alcohol functions (as in galactose) to aldehydes. A variant of the GOx from Fusarium sp. (GOxM3-5) was evolved in the laboratory to perform the oxidation of benzylic primary and secondary alcohols to the related carbonyl compounds. We recently discovered that GOxM3-5 can also promiscuously catalyze the oxidation of aldehydes to nitriles in the presence of ammonia and air. Furthermore, we developed a concurrent, two-step biocatalytic cascade that exploits both the natural and promiscuous catalytic activities of GOx in order to perform the one-pot oxidation of primary alcohols into their corresponding nitriles. Finally, the biocatalytic process was applied for the preparative scale synthesis of 2’-fluorobenzonitrile from 2’-fluorobenzyl alcohol.
*Asymmetric reductive amination of ketones catalysed by amine dehydrogenases*
Tseliou, V.; Böhmer, W.; Corrado, M. L.; Masman, M. F.; Knaus, T.; Mutti, F. G.; Chapter 5.6 in Applied Biocatalysis: The Chemist's Enzyme Toolkit, (Eds. Whittall, J.; Sutton P.W.), John Wiley & Sons, Inc, 2020, pages 221-231. ISBN: 978-1-119-48701-2
moreAmine dehydrogenases (AmDHs) are NADH/NADPH dependent enzymes that catalyze the reversible conversion of ketones and aldehydes into enantiomerically pure amines at the sole expense of ammonia and a hydride source. The latter is required for cofactor recycling and can usually be formate or glucose. A second enzyme such as a formate dehydrogenase (FDH) or a glucose dehydrogenase (GDH) catalyzes the cofactor recycling. The AmDH-FDH (or GDH) system offers elevated atom economy, as the buffer of the reaction (HCOONH4) directly provides the source of nitrogen and the reducing equivalents. Thus, only catalytic amount of NAD(P)H is needed while carbonate and water are the by-products.
Fourteen members of AmDHs are currently available. They have been created either by enzyme engineering starting from L-Amino acid dehydrogenases (L-AADHs) or discovered as native (nat-AmDHs) using metagenomic data. To date, the substrate scope of these AmDHs already covers a significant range of structurally diverse carbonyl compounds, thus giving access to approximately eighty α-chiral amines in high optical purity. This section describes the detailed procedure for the preparation and application of four engineered AmDHs: 1) Bb-PhAmDH engineered from Bacillus badius phenylalanine dehydrogenase; 2) cFL1-AmDH, a chimera obtained by domain shuffling from two first generation AmDH variants; 3) Rs-PheAmDH engineered from Rhodococcus sp. M4 phenylalanine dehydrogenase; and 4) LE-AmDH-v1 engineered from Geobacillus sterothemophilus ε-deaminating L-Lysine dehydrogenase. The enzymes can be applied in tandem with FDH from Candida boidinii for the reductive amination of carbonyl compounds. Overall, these AmDHs can give access to R-configured amines.
*Hydrogen-borrowing conversion of alcohols into optically active primary amines by combination of alcohol dehydrogenases and amine dehydrogenases*
Corrado, M. L.; Tseliou, V.; Houwman, J. A.; Böhmer, W.; Vilím, J.; Masman, M. F.; Knaus, T.; Mutti, F. G.; Chapter 11.8, in Applied Biocatalysis: The Chemist's Enzyme Toolkit (Eds. Whittall, J.; Sutton P.W.), John Wiley & Sons, Inc, 2020, pages 455-468. ISBN: 978-1-119-48701-2
moreThe direct amination of an alcohol possesses the inherent advantage that the reagent and product are in the same oxidation state; therefore, external redox equivalents are not required. However, the first challenge is to perform a tandem-process in which an oxidative and a reductive step are running simultaneously. The second challenge is to perform a redox-neutral process in which the electrons liberated in the first oxidative step are quantitatively recycled and consumed in the subsequent reductive step. Such a process is defined as “hydrogen-borrowing.” This section is particularly focused on the hydrogen-borrowing conversion of racemic alcohols into optically active α-chiral amines. In this process, the hydride released in the first step—the oxidation of the alcohol to the ketone catalyzed by alcohol dehydrogenase (ADH)—is consumed in the second step, which is the reductive amination of the ketone catalyzed by amine dehydrogenase (AmDH). The redox self-sufficient cycle uses ammonium ion/ammonia as a nitrogen source and generates only water as a by-product. Moreover, the cascade requires only catalytic amounts of β-nicotinamide adenine dinucleotide (NAD+) coenzyme to shuttle the hydride within the two steps.
These procedures report the use of two ADHs and two AmDHs, namely ADH from Aromatoleum aromaticum (AA-ADH), LBv-ADH variant from Lactobacillus brevis, Bb-PhAmDH variant K78S-N277L from Bacillus badius, and cFL1-AmDH chimeric variant. The method has been successfully applied for the asymmetric amination of a panel of racemic secondary alcohols. Specifically, the procedures describe hydrogen borrowing amination of racemic alcohols by:
1) using isolated enzymes in solution;
2) using co-immobilized enzymes;
3) using resting E. coli cells co-expressing the enzymes.
*Biocatalyzed Aerobic Oxidation Reaction*
Vilím, J.; Knaus, T.; Mutti, F. G. Chapter 6, in Catalytic Aerobic Oxidations, (Ed. Mejía, E.), RSC Catalytic Series, RSC Publishing, Cambridge (UK), 2020, pages 131-180. Online ISBN: 978-1-83916-034-9, Print ISBN: 978-1-78801-720-6
moreOxidoreductase enzymes enable a large variety of oxidation and oxyfunctionalization reactions at the expense of molecular oxygen, which is most commonly used in the form of air and at atmospheric pressure. Over the past decade, the number of available enzymes and methodologies enabling the performance of these types of reactions has increased significantly, thereby greatly complicating the navigation of the biochemical landscape of aerobic enzymatic reactions. This chapter provides an overview of bio-catalytic reactions that utilize dioxygen as a final electron acceptor or hydroxylating agent with a focus on more mature processes that allow at least gram-scale biotransformations with significant chemical turnovers, thus demonstrating practical applicability in organic synthesis. The described aerobic bio-catalytic reactions comprise: (a) carbon–hydrogen hydroxylation or halogenation; (b) mono- or di-hydroxylation as well as epoxidation or cleavage of alkenes; (c) Baeyer–Villiger oxidation; (d) oxidation of alcohols or aldehydes; oxidative decarboxylation; (e) oxidation of amines or imines; oxidation of organosulfur, organoboron or organoselenium compounds; and (f) oxidative carbon–carbon bond formation. Additionally, this chapter provides brief and selected mechanistic insights into the enzyme classes (i.e., oxygenases, oxidases, and dehydrogenases) that catalyze these biochemical transformations with often excellent chemo-, regio- and stereoselectivities.
*Enhancing the Applicability of the Bioamination of Ketones in Neat Organic Solvents using ω-Transaminases as Immobilized Biocatalysts in Continuous Flow*
Böhmer, W.; Volkov, A.; Engelmark Cassimjee, K.; Mutti, F. G.* Adv. Synth. Catal., 2020, 362, 1858–1867. Front cover article of issue issue 9 (vol. 362). DOI: 10.1002/adsc.201901274
more
Compared with biocatalysis in aqueous media, the use of enzymes in neat organic solvents enables increased solubility of hydrophobic substrates and can lead to more favorable thermodynamic equilibria, avoidance of possible hydrolytic side reactions and easier product recovery. ω‐Transaminases from Arthrobacter sp. (AsR−ωTA) and Chromobacterium violaceum (Cv−ωTA) were immobilized on controlled porosity glass metal‐ion affinity beads (EziG) and applied in neat organic solvents for the amination of 1‐phenoxypropan‐2‐one with 2‐propylamine. The reaction system was investigated in terms of type of carrier material, organic solvents and reaction temperature. Optimal conditions were found with more hydrophobic carrier materials and toluene as reaction solvent. The system's water activity (aw) was controlled via salt hydrate pairs during both the biocatalyst immobilization step and the progress of the reaction in different non‐polar solvents. Notably, the two immobilized ωTAs displayed different optimal values of aw, namely 0.7 for EziG3−AsR−ωTA and 0.2 for EziG3−Cv−ωTA. In general, high catalytic activity was observed in various organic solvents even when a high substrate concentration (450–550 mM) and only one equivalent of 2‐propylamine were applied. Under batch conditions, a chemical turnover (TTN) above 13000 was obtained over four subsequent reaction cycles with the same batch of EziG‐immobilized ωTA. Finally, the applicability of the immobilized biocatalyst in neat organic solvents was further demonstrated in a continuous flow packed‐bed reactor. The flow reactor showed excellent performance without observable loss of enzymatic catalytic activity over several days of operation. In general, ca. 70% conversion was obtained in 72 hours using a 1.82 mL flow reactor and toluene as flow solvent, thus affording a space‐time yield of 1.99 g L−1 h−1. Conversion reached above 90% when the reaction was run up to 120 hours.
*Kinetic Resolution of Racemic Primary Amines using Geobacillus stearothermophilus Amine Dehydrogenase variant*
Tseliou, V.; Knaus, T.; Vilím, J.; Masman, M. F.; Mutti, F. G.* ChemCatChem, 2020, 12, 2184-2188. DOI: 10.1002/cctc.201902085
more
A NADH-dependent engineered amine dehydrogenase from Geobacillus stearothermophilus (LE-AmDH-v1) was applied together with a NADH-oxidase from Streptococcus mutans (NOx) for the kinetic resolution of pharmaceutically relevant racemic α-chiral primary amines. The reaction conditions (e. g., pH, temperature, type of buffer) were optimised to yield S-configured amines with up to >99 % ee.
*Parallel Interconnected Kinetic Asymmetric Transformation (PIKAT) with an Immobilized ω-Transaminase in Neat Organic Solvent*
Böhmer, W.; Koenekoop, L.; Simon, T.; Mutti, F. G.* Molecules, 2020, 25, 2140. DOI: 10.3390/molecules25092140
more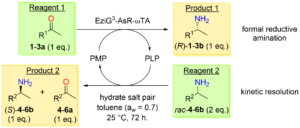
Comprising approximately 40% of the commercially available optically active drugs, α-chiral amines are pivotal for pharmaceutical manufacture. In this context, the enzymatic asymmetric amination of ketones represents a more sustainable alternative than traditional chemical procedures for chiral amine synthesis. Notable advantages are higher atom-economy and selectivity, shorter synthesis routes, milder reaction conditions and the elimination of toxic catalysts. A parallel interconnected kinetic asymmetric transformation (PIKAT) is a cascade in which one or two enzymes use the same cofactor to convert two reagents into more useful products. Herein, we describe a PIKAT catalyzed by an immobilized ω-transaminase (ωTA) in neat toluene, which concurrently combines an asymmetric transamination of a ketone with an anti-parallel kinetic resolution of an amine racemate. The applicability of the PIKAT was tested on a set of prochiral ketones and racemic α-chiral amines in a 1:2 molar ratio, which yielded elevated conversions (up to >99%) and enantiomeric excess (ee, up to >99%) for the desired products. The progress of the conversion and ee was also monitored in a selected case. This is the first report of a PIKAT using an immobilized ωTA in a non-aqueous environment.
2019
*Regio- and stereoselective multi-enzymatic aminohydroxylation of β-methylstyrene using dioxygen, ammonia and formate*
Corrado, M.L.; Knaus, T.; Mutti, F.G.* Green Chemistry, 2019, 21(23), 6246-6251, DOI: 10.1039/C9GC03161H
more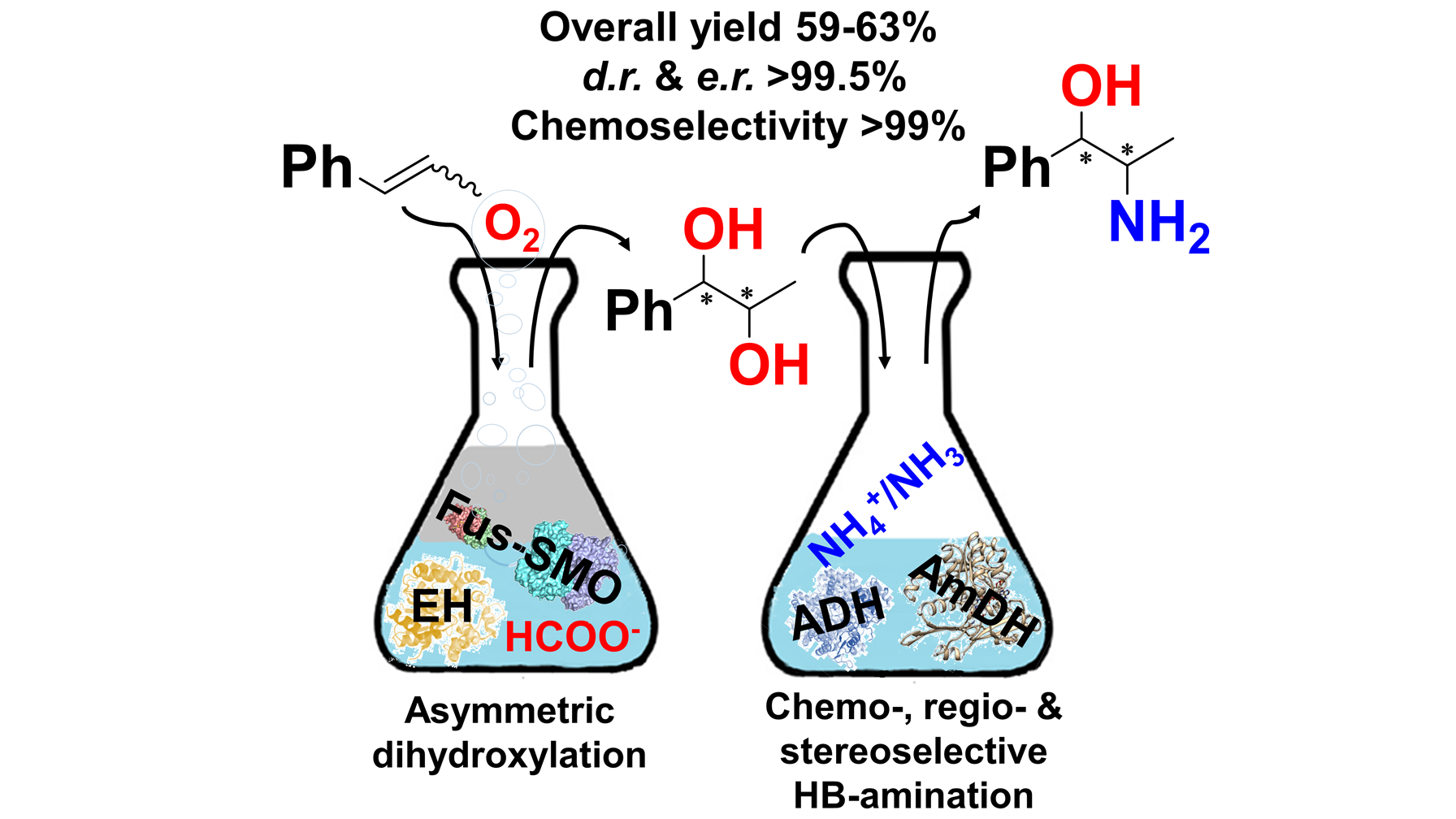
We report an enzymatic route for the formal regio- and stereoselective aminohydroxylation of β-methylstyrene that consumes only dioxygen, ammonia and formate; carbonate is the by-product. The biocascade entails highly selective epoxidation, hydrolysis and hydrogen-borrowing alcohol amination. Thus, β-methylstyrene was converted into 1R,2R and 1S,2R-phenylpropanolamine in 59–63% isolated yields, and up to >99.5 : <0.5 dr and er.
*Synthesis of enantiomerically pure alcohols and amines via biocatalytic deracemisation methods*
Musa, M.*, Hollmann, F.*; Mutti, F.G.* Catal. Sci. Technol., 2019, Advance Article. DOI: 10.1039/C9CY01539F
more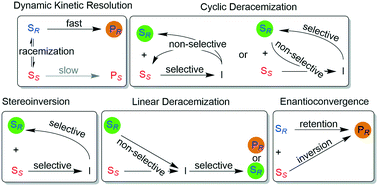
Deracemisation via chemo-enzymatic or multi-enzymatic approaches is the optimum substitute for kinetic resolution, which suffers from the limitation of a theoretical maximum 50% yield albeit high enantiomeric excess is attainable. This review covers the recent progress in various deracemisation approaches applied to the synthesis of enantiomerically pure alcohols and amines, such as (1) dynamic kinetic resolution, (2) cyclic deracemisation, (3) linear deracemisation (including stereoinversion) and (4) enantioconvergent methods.
*Generation of amine dehydrogenases with increased catalytic performance and substrate scope from ε-deaminating L-Lysine dehydrogenase*
Tseliou, V., Knaus, T.*; Masman, M.F.; Corrado, M.L.; Mutti, F.G.* Nature Communications, 2019, 10: 3717. DOI: 10.1038/s41467-019-11509-x
more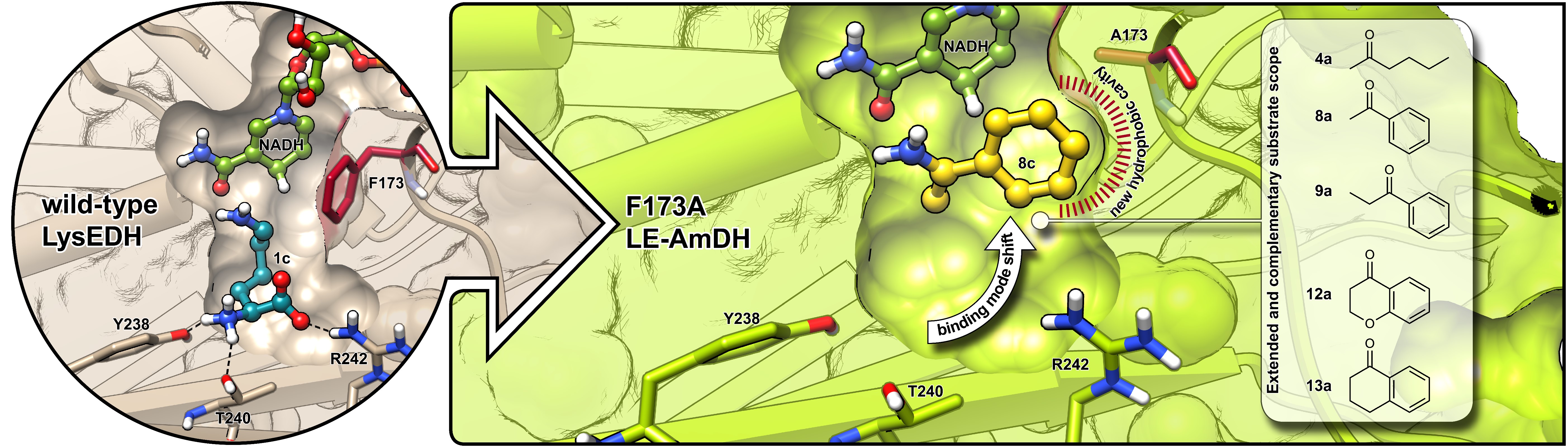
Amine dehydrogenases (AmDHs) catalyse the conversion of ketones into enantiomerically pure amines at the sole expense of ammonia and hydride source. Guided by structural information from computational models, we create AmDHs that can convert pharmaceutically relevant aromatic ketones with conversions up to quantitative and perfect chemical and optical purities. These AmDHs are created from an unconventional enzyme scaffold that apparently does not operate any asymmetric transformation in its natural reaction. Additionally, the best variant (LE-AmDH-v1) displays a unique substrate-dependent switch of enantioselectivity, affording S- or R-configured amine products with up to >99.9% enantiomeric excess. These findings are explained by in silico studies. LE-AmDH-v1 is highly thermostable (Tm of 69 °C), retains almost entirely its catalytic activity upon incubation up to 50 °C for several days, and operates preferentially at 50 °C and pH 9.0. This study also demonstrates that product inhibition can be a critical factor in AmDH-catalysed reductive amination.
*Efficient synthesis of enantiopure amines from alcohols using resting E. coli cells and ammonia*
Houwman, J. A., Knaus, T.; Costa, M.; Mutti, F.G.* Green Chemistry, 2019, 21(14): 3846-3857. DOI: 10.1039/C9GC01059A
more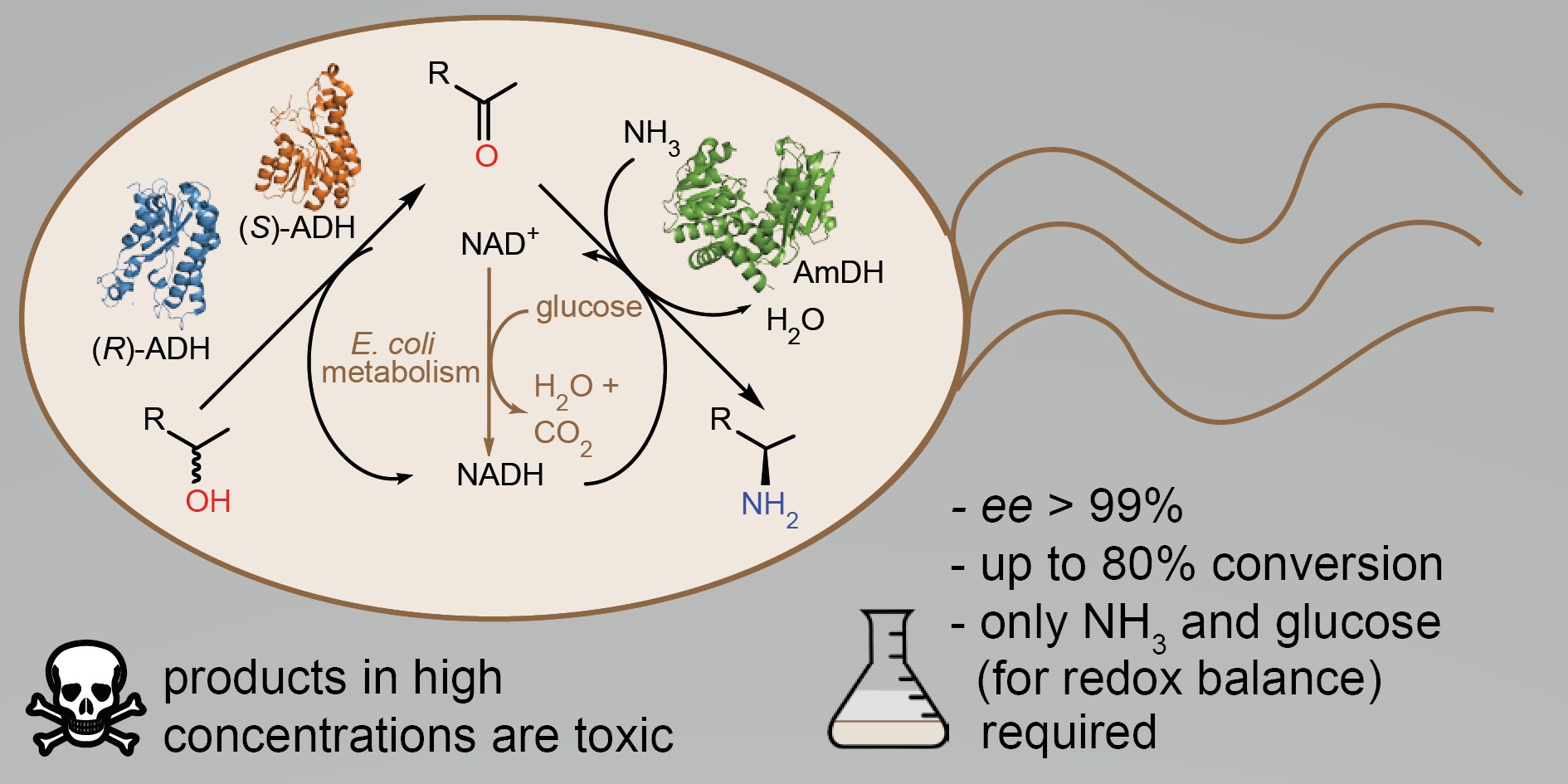
α-Chiral amines are pivotal building blocks for chemical manufacturing. Stereoselective amination of alcohols is receiving increased interest due to its higher atom-efficiency and overall improved environmental footprint compared with other chemocatalytic and biocatalytic methods. We previously developed a hydrogen-borrowing amination by combining an alcohol dehydrogenase (ADH) with an amine dehydrogenase (AmDH) in vitro. Herein, we implemented the ADH-AmDH bioamination in resting Escherichia coli cells for the first time. Different genetic constructs were created and tested in order to obtain balanced expression levels of the dehydrogenase enzymes in E. coli. Using the optimized constructs, the influence of several parameters towards the productivity of the system were investigated such as the intracellular NAD+/NADH redox balance, the cell loading, the survival rate of recombinant E. coli cells, the possible toxicity of the components of the reaction at different concentrations and the influence of different substrates and cosolvents. In particular, the cofactor redox-balance for the bioamination was maintained by the addition of moderate and precise amounts of glucose. Higher concentrations of certain amine products resulted in toxicity and cell death, which could be alleviated by the addition of a co-solvent. Notably, amine formation was consistent using several independently grown E. coli batches. The optimized E. coli/ADH-AmDH strains produced enantiopure amines from the alcohols with up to 80% conversion and a molar productivity up to 15 mM. Practical applicability was demonstrated in a gram-scale biotransformation. In summary, the present E. coli-ADH-AmDH system represents an important advancement towards the development of ‘green’, efficient and selective biocatalytic processes for the amination of alcohols.
*A Photo-Enzymatic Cascade to Transform Racemic Alcohols into Enantiomerically Pure Amines*
Gacs, J.; Zhang, W.; Knaus, T.; Mutti, F.G.; Arends, I.W.; Hollmann, F.*; Catalysts, 2019, 9 (4) , 305. DOI: 10.3390/catal9040305
more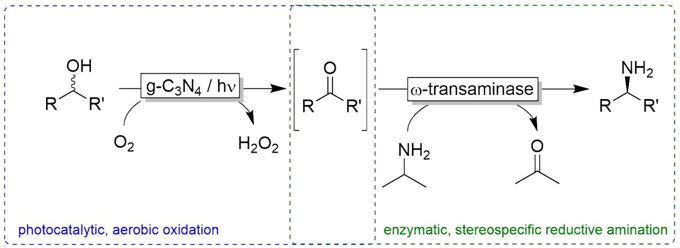
The consecutive photooxidation and reductive amination of various alcohols in a cascade reaction were realized by the combination of a photocatalyst and several enzymes. Whereas the photocatalyst (sodium anthraquinone-2-sulfonate) mediated the light-driven, aerobic oxidation of primary and secondary alcohols, the enzymes (various ω-transaminases) catalyzed the enantio-specific reductive amination of the intermediate aldehydes and ketones. The system worked in a one-pot one-step fashion, whereas the productivity was significantly improved by switching to a one-pot two-step procedure. A wide range of aliphatic and aromatic compounds was transformed into the enantiomerically pure corresponding amines via the photo-enzymatic cascade.
*Mechanistic Insight into the Catalytic Promiscuity of Amine Dehydrogenases: Asymmetric Synthesis of Secondary and Primary Amines*
Tseliou, V.; Masman M.F.; Böhmer W.; Knaus, T.; Mutti, F. G.*; ChemBioChem, 2019, 20, 800. DOI: 10.1002/cbic.201800626
more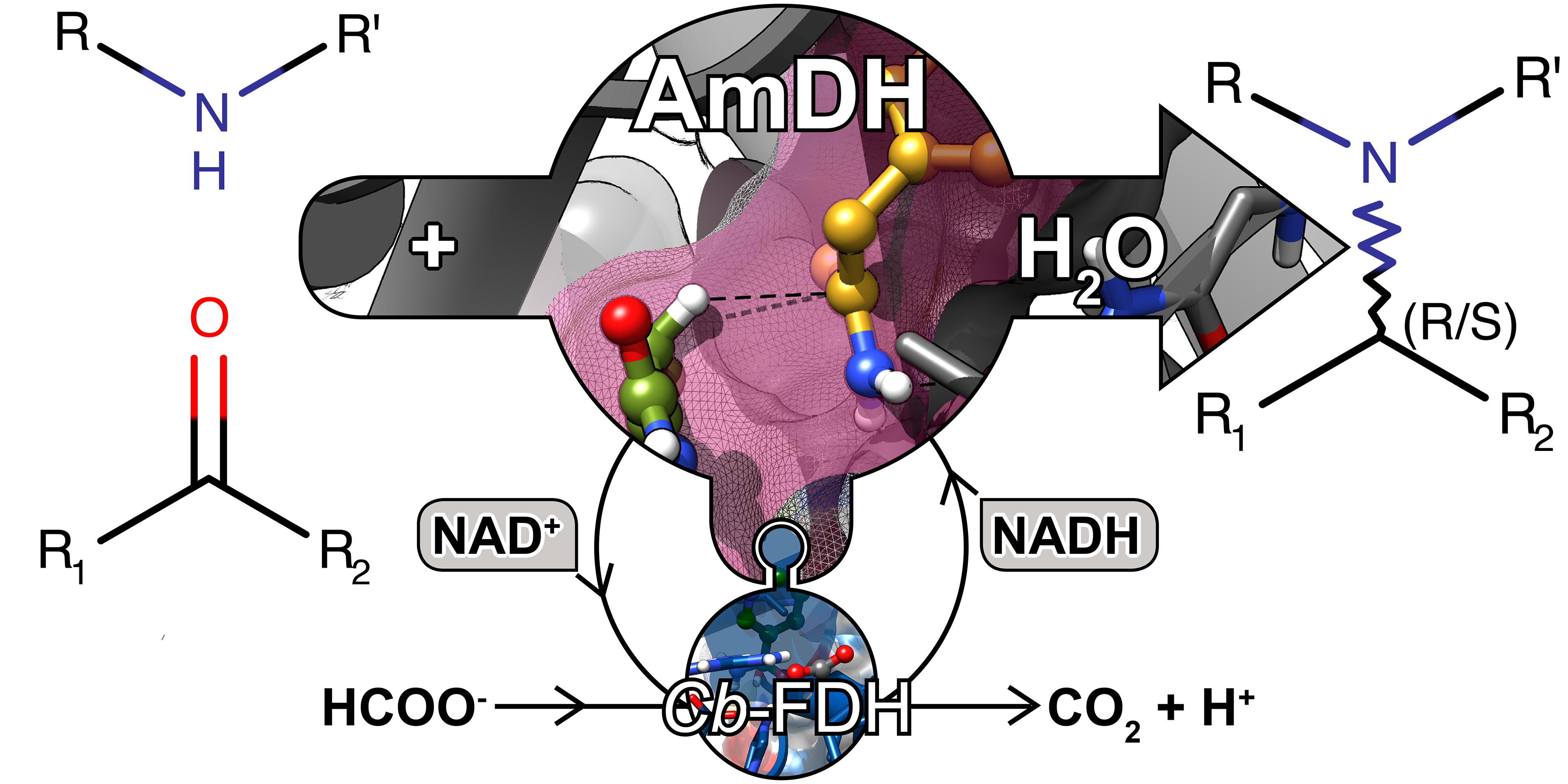
Biocatalytic asymmetric aminations of ketones using amine dehydrogenases (AmDHs) or transaminases, are efficient methods for the synthesis of α‐chiral primary amines. A major challenge is the extension of the amination to the synthesis of secondary and tertiary amines. Herein, we show for the first time that AmDHs are capable of accepting other amine donors, hence giving access to enantioenriched secondary amines with conversions up to 43%. Surprisingly, in several cases, we observed the promiscuous formation of enantiopure primary amines along with the expected secondary amines. By conducting practical laboratory experiments and computational experiments, we propose that the promiscuous formation of primary amines along with secondary amines is due to an unprecedented nicotinamide (NAD)‐dependent formal transamination catalysed by AmDHs. In nature, this type of mechanism is commonly performed by pyridoxal 5’‐phosphate (PLP) aminotransferase and not by dehydrogenases. Finally, we propose a catalytic pathway that rationalises the promiscuous NAD‐dependent formal transamination activity and explains the formation of the observed mixture of products. This work increases our understanding on the catalytic mechanism of NAD‐dependent aminating enzymes such as AmDHs and will aid further research on the rational engineering of oxidoreductases for the synthesis of α‐chiral secondary and tertiary amines.
*Highly efficient production of chiral amines in batch and continuous flow by immobilized ω-transaminases on controlled porosity glass metal-ion affinity carrier*
Böhmer, W., Knaus, T., Volkov, A., Slot, T. K., Shiju N. R., Engelmark Cassimjee, K., Mutti, F. G.*; Journal of Biotechnology, 2019, 291, 52-60. DOI: 10.1016/j.jbiotec.2018.12.001
more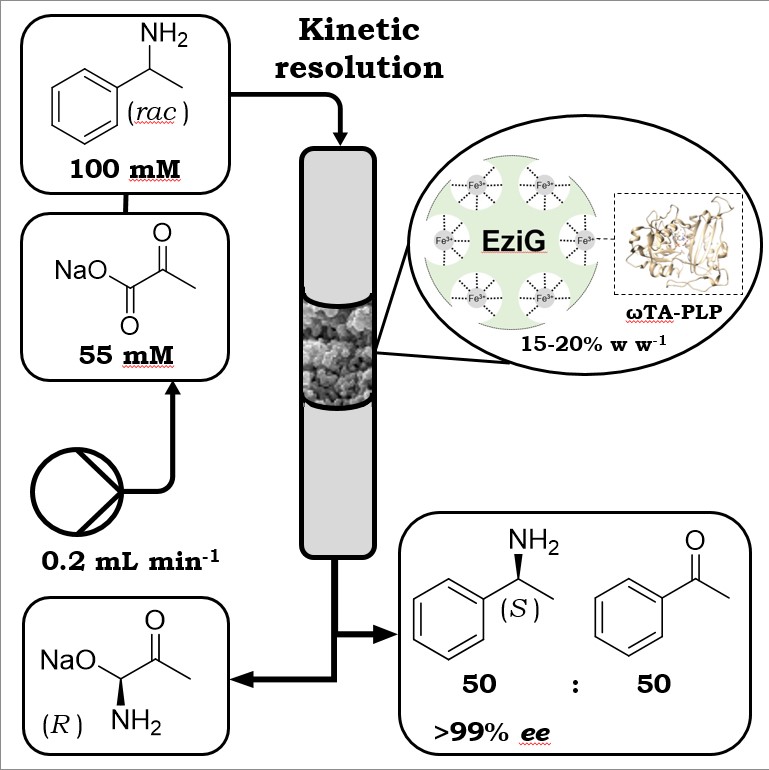
In this study, two stereocomplementary ω-transaminases from Arthrobacter sp. (AsR-ωTA) and Chromobacterium violaceum (Cv-ωTA) were immobilized via iron cation affinity binding onto polymer-coated controlled porosity glass beads (EziG™). The immobilization procedure was studied with different types of carrier materials and immobilization buffers of varying compositions, concentrations, pHs and cofactor (PLP) concentrations. Notably, concentrations of PLP above 0.1 mM were correlated with a dramatic decrease of the immobilization yield. The highest catalytic activity, along with quantitative immobilization, was obtained in MOPS buffer (100 mM, pH 8.0, PLP 0.1 mM, incubation time 2 h). Leaching of the immobilized enzyme was not observed within 3 days of incubation. EziG-immobilized AsR-ωTA and Cv-ωTA retained elevated activity when tested for the kinetic resolution of rac-α-methylbenzylamine (rac-α-MBA) in single batch experiments. Recycling studies demonstrated that immobilized EziG3-AsR-ωTA could be recycled for at least 16 consecutive cycles (15 min per cycle) and always affording quantitative conversion (TON ca. 14,400). Finally, the kinetic resolution of rac-α-MBA with EziG3-AsR-ωTA was tested in a continuous flow packed-bed reactor (157 μL reactor volume), which produced more than 5 g of (S)-α-MBA (>49% conversion, >99% ee) in 96 h with no detectable loss of catalytic activity. The calculated TON was more than 110,000 along with a space-time yield of 335 g L−1 h−1.
*Hydrolases-mediated transformation of oleuropein into demethyloleuropein*
Cariati, L.; Oliverio, M.*; Mutti, F. G.; Bonacci, S.; Knaus, T.; Costanzo, P.; Procopio, A.; Bioorganic Chemistry, 2019, 84, 384-388. DOI: 10.1016/j.bioorg.2018.12.005
more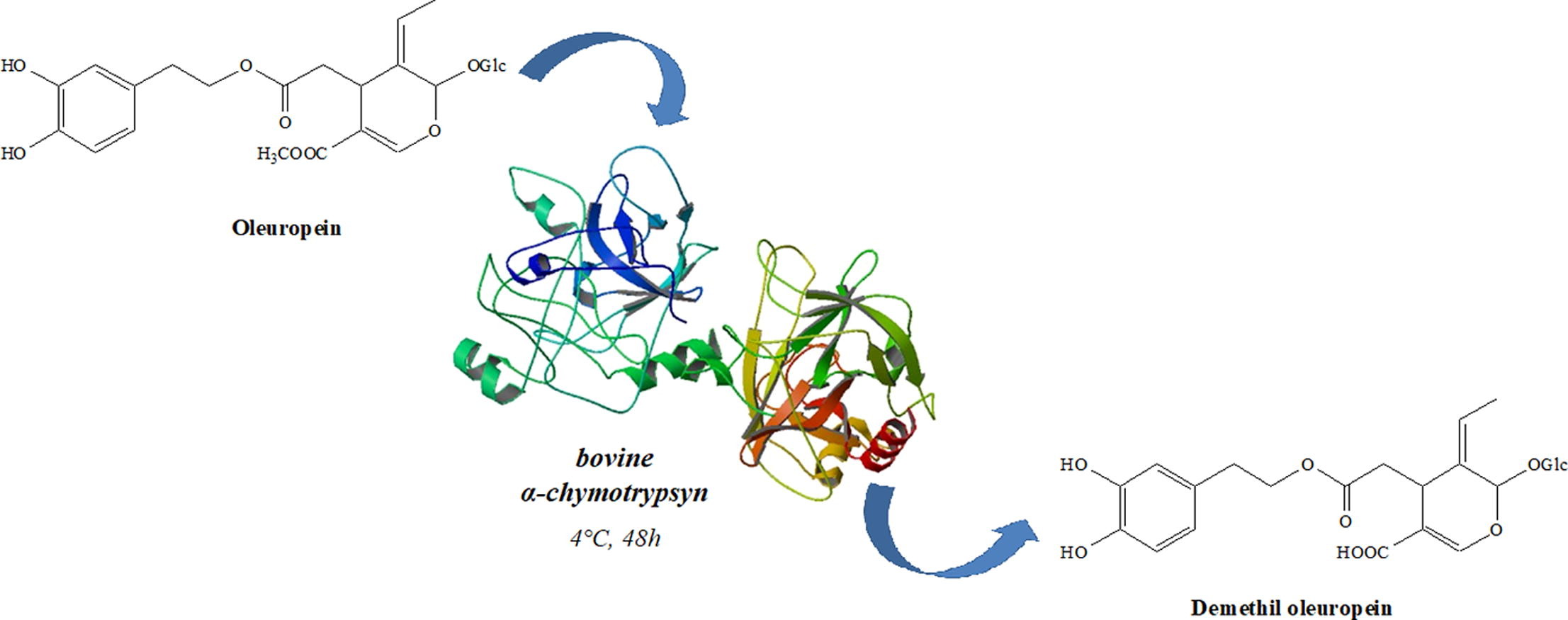
Phenolic compounds present in extra virgin olive oil have recently attracted considerable attention due to their pharmacological activities. Among them oleacein (3,4-DHPEA-EDA), structurally related to oleochantal (4-HPEA-EDA), is one of the most studied. 3,4-DHPEA-EDA has been synthesized through decarboxylation of demethyloleuropein catalyzed by Er(OTf)3. Demethyloleuropein is extracted from black olives drupes in very limited amounts and only in particular periods of the year. The availability of demethyloleuropein could be increased by a selective hydrolysis of the methyl ester moiety of oleuropein, a secoiridoid present in large amount in olive leaves. In this work we describe a new enzymatic method for carrying out a selective hydrolysis of oleuropein via the screening of a panel of hydrolases (lipases, esterases and proteases). Among all the enzymes tested the best results was obtained using α-chymotrypsyn from bovine pancreas as biocatalyst, thus revealing a classic example of catalytic enzyme promiscuity.
2018
*Catalytic Promiscuity of Galactose Oxidase: A Mild Synthesis of Nitriles from Alcohols, Air, and Ammonia*
Vilim, J.; Knaus, T.; Mutti, F. G.*; Angew. Chem. Int. Ed., 2018, 57, 14240-14244. DOI: 10.1002/anie.201809411
more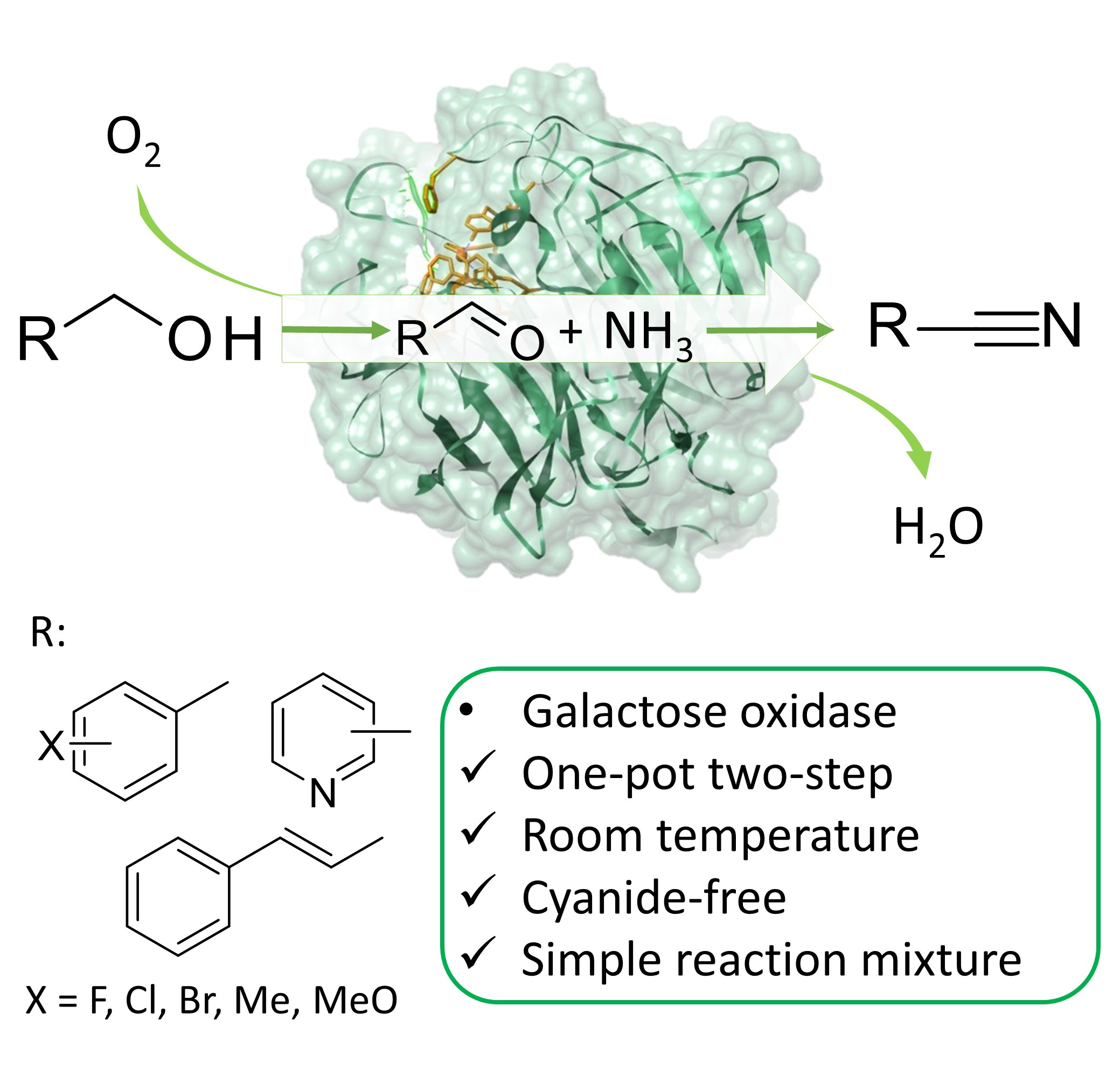
We report an unprecedented catalytically promiscuous activity of the copper‐dependent enzyme galactose oxidase. The enzyme catalyses the one‐pot conversion of alcohols into the related nitriles under mild reaction conditions in ammonium buffer, consuming ammonia as the source of nitrogen and dioxygen (from air at atmospheric pressure) as the only oxidant. Thus, this green method does not require either cyanide salts, toxic metals, or undesired oxidants in stoichiometric amounts. The substrate scope of the reaction includes benzyl and cinnamyl alcohols as well as 4‐ and 3‐pyridylmethanol, giving access to valuable chemical compounds. The oxidation proceeds through oxidation from alcohol to aldehyde, in situ imine formation, and final direct oxidation to nitrile.
*A biocatalytic method for the chemoselective aerobic oxidation of aldehydes to carboxylic acids*
Knaus T., Tseliou V., Humphreys L.D., Scrutton N.S., Mutti, F.G.* Green Chemistry, 2018, 20, 3931-3943. DOI: 10.1039/C8GC01381K
more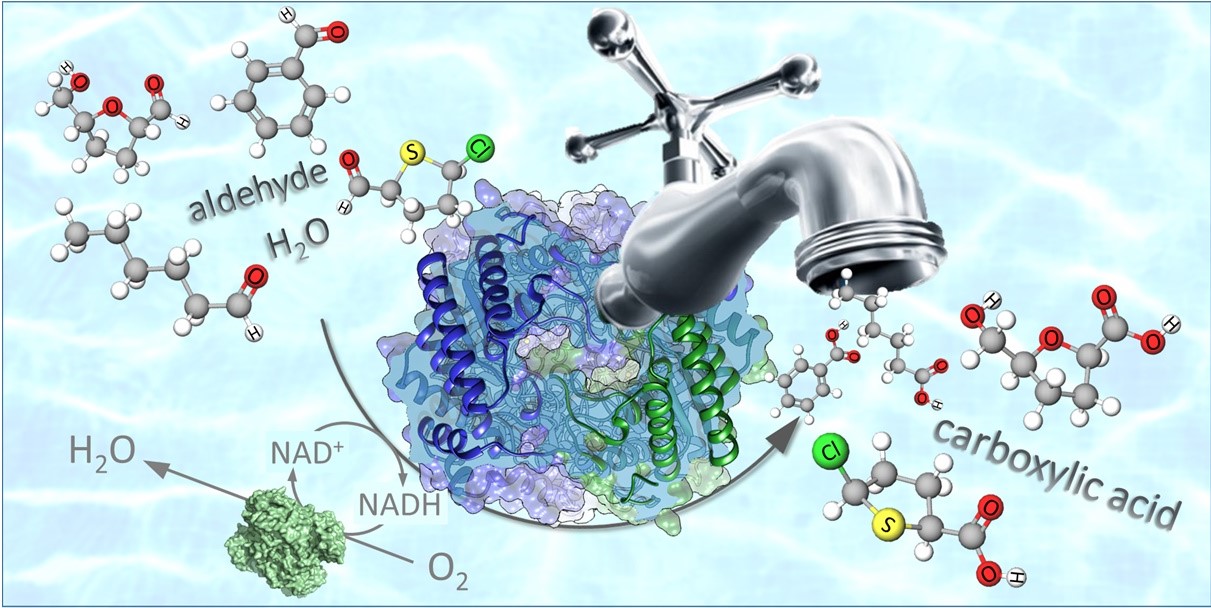
Herein, we present a study on the oxidation of aldehydes to carboxylic acids using three recombinant aldehyde dehydrogenases (ALDHs). The ALDHs were used in purified form with a nicotinamide oxidase (NOx), which recycles the catalytic NAD+ at the expense of dioxygen (air at atmospheric pressure). The reaction was studied also with lyophilised whole cell as well as resting cell biocatalysts for more convenient practical application. The optimised biocatalytic oxidation runs in phosphate buffer at pH 8.5 and at 40 °C. From a set of sixty-one aliphatic, aryl-aliphatic, benzylic, hetero-aromatic and bicyclic aldehydes, fifty were converted with elevated yield (up to >99%). The exceptions were a few ortho-substituted benzaldehydes, bicyclic heteroaromatic aldehydes and 2-phenylpropanal. In all cases, the expected carboxylic acid was shown to be the only product (>99% chemoselectivity). Other oxidisable functionalities within the same molecule (e.g. hydroxyl, alkene, and heteroaromatic nitrogen or sulphur atoms) remained untouched. The reaction was scaled for the oxidation of 5-(hydroxymethyl)furfural (2 g), a bio-based starting material, to afford 5-(hydroxymethyl)furoic acid in 61% isolated yield. The new biocatalytic method avoids the use of toxic or unsafe oxidants, strong acids or bases, or undesired solvents. It shows applicability across a wide range of substrates, and retains perfect chemoselectivity. Alternative oxidisable groups were not converted, and other classical side-reactions (e.g. halogenation of unsaturated functionalities, Dakin-type oxidation) did not occur. In comparison to other established enzymatic methods such as the use of oxidases (where the concomitant oxidation of alcohols and aldehydes is common), ALDHs offer greatly improved selectivity.
*A Chimeric Styrene Monooxygenase with Increased Efficiency in Asymmetric Biocatalytic Epoxidation*
Corrado M.L., Knaus T., Mutti F.G.* ChemBioChem, 2018, 19, 679-686. DOI: 10.1002/cbic.201700653
more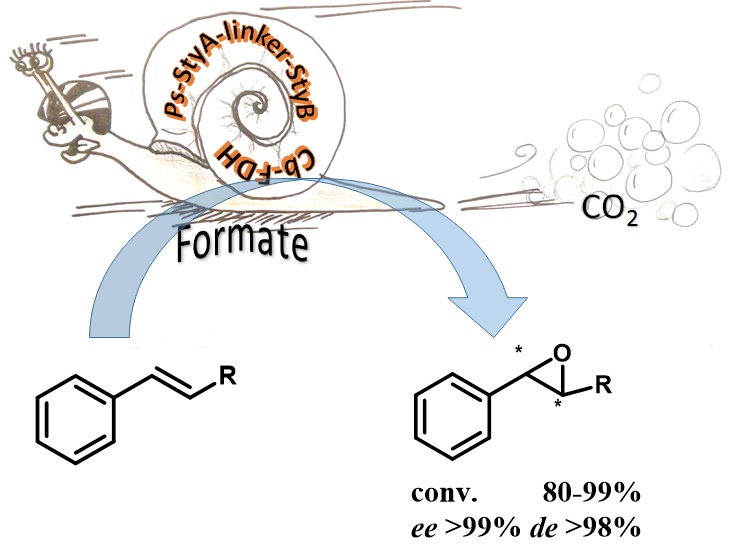
The styrene monooxygenase (SMO) system from Pseudomonas sp. consists of two enzymes (StyA and StyB). StyB catalyses the reduction of FAD at the expense of NADH. After the transfer of FADH2 from StyB to StyA, reaction with O2 generates FAD-OOH, which is the epoxidising agent. The wastage of redox equivalents due to partial diffusive transfer of FADH2 , the insolubility of recombinant StyB and the impossibility of expressing StyA and StyB in a 1:1 molar ratio reduce the catalytic efficiency of the natural system. Herein we present a chimeric SMO (Fus-SMO) that was obtained by genetic fusion of StyA and StyB through a flexible linker. Thanks to a combination of: 1) balanced and improved expression levels of reductase and epoxidase units, and 2) intrinsically higher specific epoxidation activity of Fus-SMO in some cases, Escherichia coli cells expressing Fus-SMO possess about 50% higher activity for the epoxidation of styrene derivatives than E. coli cells coexpressing StyA and StyB as discrete enzymes. The epoxidation activity of purified Fus-SMO was up to three times higher than that of the two-component StyA/StyB (1:1, molar ratio) system and up to 110 times higher than that of the natural fused SMO. Determination of coupling efficiency and study of the influence of O2 pressure were also performed. Finally, Fus-SMO and formate dehydrogenase were coexpressed in E. coli and applied as a self-sufficient biocatalytic system for epoxidation on greater than 500 mg scale.
*Hydrogen-Borrowing Alcohol Bioamination with Coimmobilized Dehydrogenases*
Böhmer W., Knaus T., Mutti F.G.*; ChemCatChem, 2018, 10, 731-735. DOI: 10.1002/cctc.201701366
more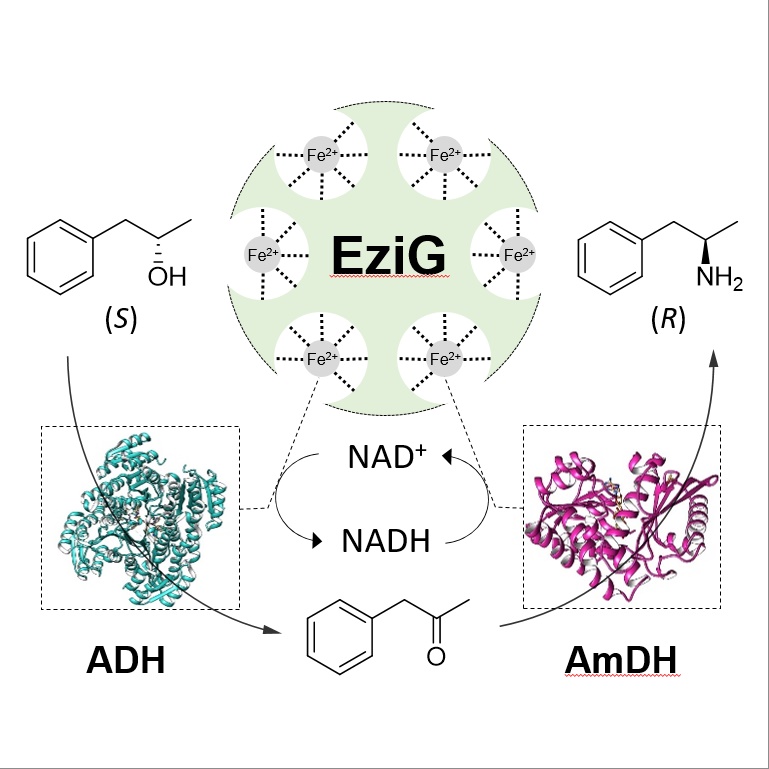
The amination of alcohols is an important transformation in chemistry. The redox‐neutral (i.e., hydrogen‐borrowing) asymmetric amination of alcohols is enabled by the combination of an alcohol dehydrogenase (ADH) with an amine dehydrogenase (AmDH). In this work, we enhanced the efficiency of hydrogen‐borrowing biocatalytic amination by co‐immobilizing both dehydrogenases on controlled porosity glass FeIII ion‐affinity beads. The recyclability of the dual‐enzyme system was demonstrated (5 cycles) with total turnover numbers of >4000 and >1000 for ADH and AmDH, respectively. A set of (S)‐configured alcohol substrates was aminated with up to 95 % conversion and >99 % ee (R). Preparative‐scale amination of (S)‐phenylpropan‐2‐ol resulted in 90 % conversion and 80 % yield of the product in 24 h.
*Selective aerobic oxidation reactions using a combination of photocatalytic water oxidation and enzymatic oxyfunctionalizations*
Zhang W., Fernández-Fueyo E., Ni Y., van Schie M., Gacs J., Renirie R., Wever R., Mutti, F.G., Rother D., Alcalde M. & Hollmann F.*; Nature Catalysis, 2018, 1, 55-62. DOI: 10.1038/s41929-017-0001-5
morePeroxygenases offer an attractive means to address challenges in selective oxyfunctionalization chemistry. Despite this, their application in synthetic chemistry remains challenging due to their facile inactivation by the stoichiometric oxidant H2O2. Often atom-inefficient peroxide generation systems are required, which show little potential for large-scale implementation. Here, we show that visible-light-driven, catalytic water oxidation can be used for in situ generation of H2O2 from water, rendering the peroxygenase catalytically active. In this way, the stereoselective oxyfunctionalization of hydrocarbons can be achieved by simply using the catalytic system, water and visible light.
2017
*In vitro biocatalytic pathway design: orthogonal network for the quantitative and stereospecific amination of alcohols*
Knaus, T.; Cariati, L.; Masman, M. F.; Mutti, F. G.*; Org. Biomol. Chem., 2017, 15, 8313-8325. DOI: 10.1039/C7OB01927K.
more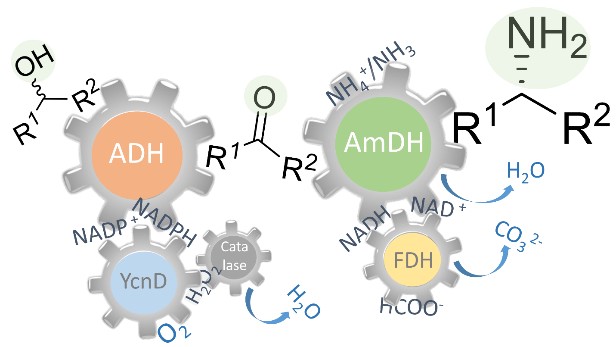
The direct and efficient conversion of alcohols into amines is a pivotal transformation in chemistry. Here, we present an artificial, oxidation–reduction, biocatalytic network that employs five enzymes (alcohol dehydrogenase, NADP-oxidase, catalase, amine dehydrogenase and formate dehydrogenase) in two concurrent and orthogonal cycles. The NADP-dependent oxidative cycle converts a diverse range of aromatic and aliphatic alcohol substrates to the carbonyl compound intermediates, whereas the NAD-dependent reductive aminating cycle generates the related amine products with >99% enantiomeric excess (R) and up to >99% conversion. The elevated conversions stem from the favorable thermodynamic equilibrium (K′eq = 1.88 × 1042 and 1.48 × 1041 for the amination of primary and secondary alcohols, respectively). This biocatalytic network possesses elevated atom efficiency, since the reaction buffer (ammonium formate) is both the aminating agent and the source of reducing equivalents. Additionally, only dioxygen is needed, whereas water and carbonate are the by-products. For the oxidative step, we have employed three variants of the NADP-dependent alcohol dehydrogenase from Thermoanaerobacter ethanolicus and we have elucidated the origin of the stereoselective properties of these variants with the aid of in silico computational models.
*Biocatalytic hydrogen-borrowing cascades*
Knaus T., Mutti F.G.*; Chim. Oggi., 2017, 35(5), 34-34. (open access upon registration) (monographic special issue)
more
The exquisite chemoselectivity and the intrinsic compatibility of enzymes have been widely exploited during the past decade for the development of multi-step biocatalytic reactions in one-pot. In this context, hydrogen-borrowing cascades permit to maximise the atom-efficiency through the internal recycling of redox equivalents, which avoids the use of additional oxidants or reductants. In this mini-review, we describe the state-of-the-art in the field of biocatalytic hydrogen-borrowing cascades and provide a future perspective for a wider implementation in organic synthesis.
*Amine dehydrogenases: efficient biocatalysts for the reductive amination of carbonyl compounds*
Knaus, T.; Böhmer, W.; Mutti, F.G.*; Green Chem., 2017, 19, 453-463. DOI: 10.1039/C6GC01987K
more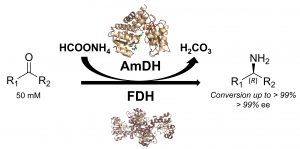
Amines constitute the major targets for the production of a plethora of chemical compounds that have applications in the pharmaceutical, agrochemical and bulk chemical industries. However, the asymmetric synthesis of α-chiral amines with elevated catalytic efficiency and atom economy is still a very challenging synthetic problem. Here, we investigated the biocatalytic reductive amination of carbonyl compounds employing a rising class of enzymes for amine synthesis: amine dehydrogenases (AmDHs). The three AmDHs from this study – operating in tandem with a formate dehydrogenase from Candida boidinii (Cb-FDH) for the recycling of the nicotinamide coenzyme – performed the efficient amination of a range of diverse aromatic and aliphatic ketones and aldehydes with up to quantitative conversion and elevated turnover numbers (TONs). Moreover, the reductive amination of prochiral ketones proceeded with perfect stereoselectivity, always affording the (R)-configured amines with more than 99% enantiomeric excess. The most suitable amine dehydrogenase, the optimised catalyst loading and the required reaction time were determined for each substrate. The biocatalytic reductive amination with this dual-enzyme system (AmDH–Cb-FDH) possesses elevated atom efficiency as it utilizes the ammonium formate buffer as the source of both nitrogen and reducing equivalents. Inorganic carbonate is the sole by-product.
2016
*Better than Nature: Nicotinamide Biomimetics That Outperform Natural Coenzymes*
Knaus, T.; Paul, C.E.; Levy, C.W.; de Vries, S.; Mutti, F.G.; Hollmann, F.; Scrutton, N.S.*; J. Am. Chem. Soc., 2016, 138 (3), 1033–1039. DOI: 10.1021/jacs.5b12252
more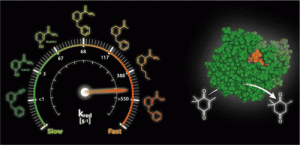
The search for affordable, green biocatalytic processes is a challenge for chemicals manufacture. Redox biotransformations are potentially attractive, but they rely on unstable and expensive nicotinamide coenzymes that have prevented their widespread exploitation. Stoichiometric use of natural coenzymes is not viable economically, and the instability of these molecules hinders catalytic processes that employ coenzyme recycling. Here, we investigate the efficiency of man-made synthetic biomimetics of the natural coenzymes NAD(P)H in redox biocatalysis. Extensive studies with a range of oxidoreductases belonging to the “ene” reductase family show that these biomimetics are excellent analogues of the natural coenzymes, revealed also in crystal structures of the ene reductase XenA with selected biomimetics. In selected cases, these biomimetics outperform the natural coenzymes. “Better-than-Nature” biomimetics should find widespread application in fine and specialty chemicals production by harnessing the power of high stereo-, regio-, and chemoselective redox biocatalysts and enabling reactions under mild conditions at low cost.
*Whole-Cell Biocatalysts for Stereoselective C−H Amination Reactions*
Both, P.; Busch, H.; Kelly, P.P.; Mutti, F.G.; Turner, N.J.; Flitsch, S.L.*; Angew. Chem. Int. Ed., 2016, 55, 1511-1513. DOI: 10.1002/anie.201510028
more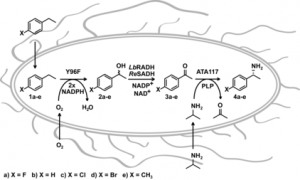
Enantiomerically pure chiral amines are ubiquitous chemical building blocks in bioactive pharmaceutical products and their synthesis from simple starting materials is of great interest. One of the most attractive strategies is the stereoselective installation of a chiral amine through C−H amination, which is a challenging chemical transformation. Herein we report the application of a multienzyme cascade, generated in a single bacterial whole-cell system, which is able to catalyze stereoselective benzylic aminations with ee values of 97.5 %. The cascade uses four heterologously expressed recombinant enzymes with cofactors provided by the host cell and isopropyl amine added as the amine donor. The cascade presents the first example of the successful de novo design of a single whole-cell biocatalyst for formal stereoselective C−H amination.
Earlier than 2015
*Conversion of alcohols to enantiopure amines through dual enzyme hydrogen-borrowing cascades*
Mutti, F.G.*; Knaus, T.; Scrutton, N.S.; Breuer, M.; Turner, N. J.*; Science, 2015, 349 (6255), 1525-1529. DOI: 10.1126/science.aac9283 (open access: http://europepmc.org/articles/PMC4883652)
more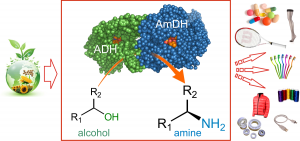
A biocatalytic hydrogen-borrowing amination of primary and secondary alcohols that allows for the efficient and environmentally benign production of enantiopure amines is presented. The method relies on a combination of two enzymes: an alcohol dehydrogenase operating in tandem with an amine dehydrogenase to aminate a structurally diverse range of aromatic and aliphatic alcohols, yielding up to 96% conversion and 99% enantiomeric excess. Primary alcohols were aminated with high conversion (up to 99%). This redox self-sufficient network possesses high atom efficiency, sourcing nitrogen from ammonium and generating water as the sole by-product.
*Asymmetric biocatalytic amination of ketones at the expense of ammonia and molecular hydrogen*
Holzer, A.K.; Hiebler, K.; Mutti, F.G.; Simon, R.C.; Lauterbach, L.; Lenz, O.; Kroutil, W.; Org. Lett., 2015, 17, 2431-2433. DOI: 10.1021/acs.orglett.5b01154
more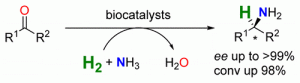
A biocatalytic system is presented for the stereoselective amination of ketones at the expense of NH3 and molecular hydrogen. By using a NAD+-reducing hydrogenase, an alanine dehydrogenase, and a suitable ω-transaminase, the R- as well as the S-enantiomer of various amines could be prepared with up to >99% ee and 98% conversion.
*Systematic methodology for the development of biocatalytic hydrogen-borrowing cascades: Application to the synthesis of chiral α-substituted carboxylic acids from α-substituted α,β-unsaturated aldehydes*
Knaus, T.; Mutti, F.G.; Humphreys, L.; Turner, N.J.; Scrutton, N.S.; Org. Biomol. Chem., 2015, 13, 223-233. DOI: 10.1039/C4OB02282C
more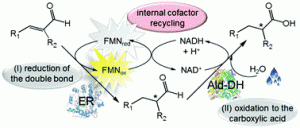
Ene-reductases (ERs) are flavin dependent enzymes that catalyze the asymmetric reduction of activated carbon-carbon double bonds. In particular, α,β-unsaturated carbonyl compounds (e.g. enals and enones) as well as nitroalkenes are rapidly reduced. Conversely, α,β-unsaturated esters are poorly accepted substrates whereas free carboxylic acids are not converted at all. The only exceptions are α,β-unsaturated diacids, diesters as well as esters bearing an electron-withdrawing group in α- or β-position. Here, we present an alternative approach that has a general applicability for directly obtaining diverse chiral α-substituted carboxylic acids. This approach combines two enzyme classes, namely ERs and aldehyde dehydrogenases (Ald-DHs), in a concurrent reductive-oxidative biocatalytic cascade. This strategy has several advantages as the starting material is an α-substituted α,β-unsaturated aldehyde, a class of compounds extremely reactive for the reduction of the alkene moiety. Furthermore no external hydride source from a sacrificial substrate (e.g. glucose, formate) is required since the hydride for the first reductive step is liberated in the second oxidative step. Such a process is defined as a hydrogen-borrowing cascade. This methodology has wide applicability as it was successfully applied to the synthesis of chiral substituted hydrocinnamic acids, aliphatic acids, heterocycles and even acetylated amino acids with elevated yield, chemo- and stereo-selectivity. A systematic methodology for optimizing the hydrogen-borrowing two-enzyme synthesis of α-chiral substituted carboxylic acids was developed. This systematic methodology has general applicability for the development of diverse hydrogen-borrowing processes that possess the highest atom efficiency and the lowest environmental impact.
*Introducing an in situ capping strategy in systems biocatalysis to access 6-aminohexanoic acid*
Sattler, J.H.; Fuchs, M.; Mutti, F.G., Grischek, B.; Engel, P., Pfeffer, J.; Woodley, J. M.; Kroutil, W.; Angew. Chem. Int. Ed., 2014, 53, 14153-14157. DOI: 10.1002/anie.201409227
more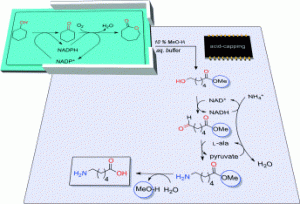
The combination of two cofactor self-sufficient biocatalytic cascade modules allowed the successful transformation of cyclohexanol into the nylon-6 monomer 6-aminohexanoic acid at the expense of only oxygen and ammonia. A hitherto unprecedented carboxylic acid capping strategy was introduced to minimize the formation of the dead-end intermediate 6-hydroxyhexanoic acid. For this purpose, the precursor ε-caprolactone was converted in aqueous medium in the presence of methanol into the corresponding methyl ester instead of the acid. Hence, it was shown for the first time that esterases - specifically horse liver esterase - can perform the selective ring-opening of ε-caprolactone with a clear preference for methanol over water as the nucleophile.
*Controlling stereoselectivity by enzymatic and chemical means to access enantiomerically pure (1S,3R)-1-benzyl-2,3-dimethyl-1,2,3,4-tetrahydroisoquinoline derivatives*
Orden, A.A.; Schrittwieser, J.H.; Resch, V.; Mutti, F.G.; Kroutil, W.; Tetrahedron: Asymmetry, 2013, 24, 744-749. DOI: 10.1016/j.tetasy.2013.05.003
more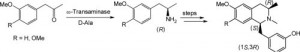
A chemoenzymatic strategy for the synthesis of enantiomerically pure novel alkaloids (1S,3R)-1-benzyl-2,3-dimethyl-1,2,3,4-tetrahydroisoquinolines is presented. The key steps are the biocatalytic stereoselective reductive amination of substituted 1-phenylpropan-2-one derivatives to yield chiral amines employing microbial ω-transaminases, and the diastereoselective reduction of a Bischler–Napieralski imine intermediate by catalytic hydrogenation in the presence of palladium on charcoal, leading exclusively to the desired cis-isomer.
*Asymmetric Preparation of prim-, sec-, and tert-amines employing selected biocatalysts*
Kroutil, W.; Fischereder, E.-M.; Fuchs, C.S.; Lerchner, H.; Mutti, F.G.; Pressnitz, D.; Rajagopalan, A.; Sattler, J.H.; Simon, R.; Siirola, E.; Org. Process Res. Dev., 2013, 17, 751-759. DOI: 10.1021/op4000237
more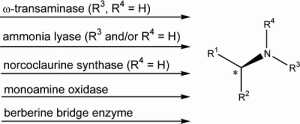
This account focuses on the application of ω-transaminases, lyases, and oxidases for the preparation of amines considering mainly work from our own lab. Examples are given to access α-chiral primary amines from the corresponding ketones as well as terminal amines from primary alcohols via a two-step biocascade. 2,6-Disubstituted piperidines, as examples for secondary amines, are prepared by biocatalytical regioselective asymmetric monoamination of designated diketones followed by spontaneous ring closure and a subsequent diastereoselective reduction step. Optically pure tert-amines such as berbines and N-methyl benzylisoquinolines are obtained by kinetic resolution via an enantioselective aerobic oxidative C–C bond formation.
*Asymmetric synthesis of 3-substituted cyclohexylamine derivatives from prochiral diketones via three biocatalytic steps*
Siirola, E.; Mutti, F.G.; Grischek, B.; Hoefler, S.F.; Fabian, W.M.F.; Grogan, G.; Kroutil, W.; Adv. Synth. Catal., 2013, 355, 1703-1708. DOI: 10.1002/adsc.201201057
more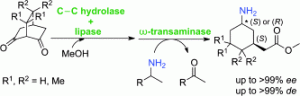
Prochiral bicyclic diketones were transformed to a single diastereomer of 3-substituted cyclohexylamine derivatives via three consecutive biocatalytic steps. The two chiral centres were set up by a C-C hydrolase (6-oxocamphor hydrolase) in the first step and by an ω-transaminase in the last step. The esterification of the intermediate keto acid was catalysed by a lipase in the second step if possible. For two substrates the C-C hydrolytic step as well as the esterification could be run simultaneously in a one-pot cascade in an organic solvent. In one example, the reaction mixture of the first two steps could be directly subjected to bio-amination in an organic solvent without the need to change the reaction medium. Depending on the choice of the ω-transaminase employed and the substrate the cis- as well as the trans-diastereomers could be obtained in optically pure forms.
*Asymmetric amination of tetralone and chromanone derivatives employing ω‑transaminases*
Pressnitz, D.; Fuchs, C.; Sattler, J.H.; Knaus, T.; Macheroux, P.; Mutti, F.G., Kroutil, W.; ACS Catalysis, 2013, 3, 555-559. DOI: 10.1021/cs400002d
more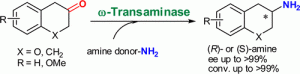
Various (S)-selective and (R)-selective ω-transaminases were investigated for the amination of 1- and 2-tetralone and derivatives as well as of 3- and 4-chromanone. All ketones tested were aminated to give the corresponding enantiopure amines (ee > 99%) employing at least one of the enzymes investigated. In most of the cases the (S)- as well as the (R)-enantiomer was obtained in optically pure form. The amination of 3-chromanone was performed on a 100 mg scale leading to optically pure (R)-3-aminochromane (ee > 99%) with complete conversion and 78% isolated yield.
*Alkene cleavage by white-rot Trametes hirsuta: Inducing enzyme activity by a fungicide*
Rajagopalan, A.; Schober, M., Mutti, F.G., Kroutil, W.; J. Mol. Catal. B: Enzym., 2013, 90, 118-122. DOI: 10.1016/j.molcatb.2013.02.002
more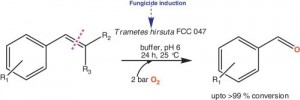
Alkene cleavage is a widely employed oxidation reaction in organic chemistry. An enzyme preparation of the wood degrading fungus Trametes hirsuta is known to cleave the C=C double bond adjacent to an aromatic ring to give the corresponding carbonyl compound at the expense of molecular oxygen as the sole oxidant. Lab-grown fungus cultures displayed varied activity and lost their alkene cleavage activity over generations of growth. t-Anethole, which is the best accepted substrate by the enzyme, is described as a major component of essential oils produced by certain plants with powerful fungicidal property. We could now show that the alkene cleaving activity was improved by the addition of the fungicide t-anethole during culture growth which represented to be an efficient method to produce cells possessing a consistent level of high alkene cleavage activity.
*Biocatalytic synthesis of enantiopure building blocks for pharmaceuticals*
Simon, R.C.; Mutti, F.G.; Kroutil, W.; Drug Discov. Today: Technol., 2013, 10, e37-44. DOI: 10.1016/j.ddtec.2012.08.002
more
Biocatalytic transformations have emerged as a viable alternative to other asymmetric chemical methods due to the intrinsic high stereoselectivity of the enzymes and the mild reaction conditions. Just a decade ago, the reaction scope of applicable biotransformations for organic synthesis was limited to a handful of reaction types. Tremendous progress has been made in the meantime so that this review presents only a small selection of the broad range of possible biotransfromations for organic synthesis available today. Lyases (hydroxynitrile lyase, aldolases) and redox enzymes like alcohol dehydrogenases, Baeyer–Villiger monooxygenase, dioxygenases, ene reductases, berberine bridge enzyme and ω-transaminases are discussed besides hydrolases.
*Asymmetric bio-amination of ketones in organic solvents*
Mutti, F.G. Kroutil W.; Adv. Synth. Catal., (2012), 354, 3409-3413. DOI: 10.1002/adsc.201200900
moreω-Transaminases, employed as a lyophilised crude cell-free extract, were successfully employed in organic solvent for the asymmetric amination of ketones without the need for immobilisation. Best activity was found for methyl tert-butyl ether (MTBE) at a water activity of 0.6. The ω-transaminases (9 different enzymes) accepted efficiently 2-propylamine as amine donor when used in the solvent, which is not the case when they are used in aqueous solution. The bio-amination in organic solvent showed several advantages such as higher reaction rates (up to 17-fold), general acceptance of 2-propylamine as amine donor, simple work-up procedure (i.e., no basification and extraction required), easy recycling of the catalyst and lack of substrate inhibition. The biocatalysts maintained their excellent stereoselectivity in MTBE allowing the preparation of optically pure amines (ee >99%) with up to >99% conversion.
*Redox self-sufficient biocatalyst network for the amination of primary alcohols*
Sattler, J.H.; Fuchs, M.; Tauber, K., Mutti, F.G., Faber, K.; Pfeffer, J.; Haas, T.; Kroutil, W.; Angew. Chem. Int. Ed., 2012, 51, 9156 – 9159. DOI: 10.1002/anie.201204683
more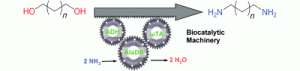
Driving the machinery: A biocatalytic redox-neutral cascade for the preparation of terminal primary amines from primary alcohols at the expense of ammonia has been established in a one-pot one-step method (see picture). Applying this artificial biocatalyst network, long-chain 1,ω-alkanediols were converted into diamines, which are building blocks for polymers, in up to 99% conversion.
*Alkene cleavage catalysed by heme and nonheme enzymes: Reaction mechanisms and biocatalytic applications*
Mutti, F.G. Bioinorganic chemistry and applications, 2012, vol 2012, Art. ID 626909, 13 pages. DOI: 10.1155/2012/626909
moreThe oxidative cleavage of alkenes is classically performed by chemical methods, although they display several drawbacks. Ozonolysis requires harsh conditions (−78 °C, for a safe process) and reducing reagents in a molar amount, whereas the use of poisonous heavy metals such as Cr, Os, or Ru as catalysts is additionally plagued by low yield and selectivity. Conversely, heme and nonheme enzymes can catalyse the oxidative alkene cleavage at ambient temperature and atmospheric pressure in an aqueous buffer, showing excellent chemo- and regioselectivities in certain cases. This paper focuses on the alkene cleavage catalysed by iron cofactor-dependent enzymes encompassing the reaction mechanisms (in case where it is known) and the application of these enzymes in biocatalysis.
*Amination of ketones by employing two new (S)-Selective ω-transaminases and the His-tagged ω-TA from Vibrio fluvialis*
Mutti, F.G.; Fuchs, C.S.; Pressnitz, D.; Turrini, N.G.; Sattler, J.H.; Lerchner, A.; Skerra, A.; Kroutil, W.; Eur. J. Org. Chem., 2012, 1003 - 1007. DOI: 10.1002/ejoc.201101476
moreTwo recently identified (S)-selective ω-transaminases (ω-TAs) that originate from Paracoccus denitrificans (Strep-PD-ωTA, cloned with an N-terminal Strep-tag II) and Pseudomonas fluorescens (PF-ωTA) were employed for the asymmetric amination of selected prochiral ketones. The substrates tested were transformed into optically pure amines (>99% ee) with high conversion (up to >99%). The ω-TAs led to higher conversion in the absence of dimethyl sulfoxide as a cosolvent than in its presence (15%, v/v). Additionally, it was shown that a His-tagged recombinant transaminase from Vibrio fluvialis (His-VF-ωTA, cloned with an N-terminal His6-tag) showed for a single substrate, ethyl acetoacetate, significantly higher stereoselectivity for the amination compared to the corresponding commercial enzyme preparation (>99 vs. 50%).
*Stereoselectivity of four (R)-selective transaminases for the asymmetric amination of ketones*
Mutti, F.G.; Fuchs C.S.; Pressnitz, D.; Sattler, J.H.; Kroutil, W.; Adv. Synth. Catal., 2011, 353, 3227 - 3233. DOI: 10.1002/adsc.201100558
more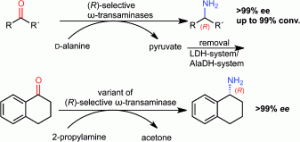
Four (R)-ω-transaminases originating from Hyphomonas neptunium (HN-ωTA), Aspergillus terreus (AT-ωTA) and Arthrobacter sp. (ArR-ωTA), as well as an evolved transaminase (ArRmut11-ωTA) were successfully employed for the amination of prochiral ketones leading to optically pure (R)-amines. The first three transaminases displayed perfect stereoselectivity for the amination of all substrates tested (ee >99%). Furthermore, the transaminase AT-ωTA led in most cases to better conversion than ArR-ωTA and HN-ωTA using D-alanine as amine donor. α-Tetralone, which was the only substrate not accepted by HN-ωTA, ArR-ωTA, and AT-ωTA, was successfully transformed with perfect enantioselectivity (ee >99%) into the corresponding optically pure amine employing the variant ArRmut11-ωTA.
*Chemoselective aerobic oxidation of 4-allylanisol by Fe(III) porphyrins in an aqueous system*
Vakuliuk, O.; Mutti, F.G.; Lara, M.; Gryko, D.T.; Kroutil, W.; Tetrahedron Lett., 2011, 52, 3555 - 3557. DOI: 10.1016/j.tetlet.2011.03.050
more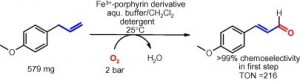
The allyl moiety of 4-allyl-anisol was oxidized in the presence of a Fe(III) porphyrin derivative to the corresponding α,β-unsaturated aldehyde in an initial oxidation step with perfect chemoselectivity. Molecular oxygen was employed as the sole environmental innocuous oxidant. The reaction was performed in an aqueous buffer/CH2Cl2 mixture using the detergent Tween 80 to homogenize the system.
*A new chiral, poly-imidazole N8-ligand and the related di- and tri-copper(II) complexes: synthesis, theoretical modelling, spectroscopic properties, and biomimetic stereoselective oxidations*
Mutti, F.G.; Gullotti, M.; Casella, L.; Santagostini, L.; Pagliarin, R.; Andersson, K.K.; Iozzi, M.F.; Zoppellaro, G.; Dalton Trans., 2011, 40, 5436 - 5457. DOI: 10.1039/C0DT00669F
more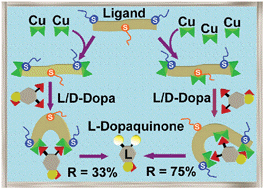
The new poly-imidazole N8 ligand (S)-2-piperazinemethanamine-1,4-bis[2-((N-(1-acetoxy-3-(1-methyl-1H-imidazol-4-yl))-2-(S)-propyl)-(N-(1-methyl-1H-imidazol-2-ylmethyl)))ethyl]-N-(phenylmethyl)-N-(acetoxy), also named (S)-Pz-(C2-(HisIm))2 (L), containing three chiral (S) centers, was obtained by a multi-step synthesis and used to prepare dinuclear [Cu2(L)]4+ and trinuclear [Cu3(L)]6+ copper(II) complexes. Low-temperature EPR experiments performed on [Cu2(L)]4+ demonstrated that the two S = ½ centers behaved as independent paramagnetic units, while the EPR spectra used to study the trinuclear copper complex, [Cu3(L)]6+, were consistent with a weakly coupled three-spin ½ system. Theoretical models for the two complexes were obtained by DFT/RI-BP86/TZVP geometry optimization, where the structural and electronic characteristics nicely supported the EPR experimental findings. In addition, the theoretical analysis unveiled that the conformational flexibility encoded in both [Cu2(L)]4+ and [Cu3(L)]6+ arises not only from the presence of several σ-bonds and the bulky residues attached to the (S)-Pz-(C2-(HisIm))2 ligand scaffold, but also from the poor coordination ability of the tertiary amino groups located in the ligand side-chains containing the imidazole units towards the copper(II) ions. Both the dinuclear and trinuclear complexes are efficient catalysts in the stereoselective oxidation of several catechols and flavonoid compounds, yielding the corresponding quinones. The structural features of the substrate–catalyst adduct intermediates were assessed by searching the conformational space of the molecule through MMFF94/Monte Carlo (MMFF94/MC) methods. The conformational flexibility of the bound ligand in the complexes proves to be beneficial for substrate binding and recognition. For the dinuclear complex, chiral recognition of the optically active substrates derives from weak electrostatic interactions between bound substrates and folded regions of the ligand scaffold. For the trinuclear complex, in the case of L/D-Dopa, the chiral recognition has a remarkable stereoselectivity index of 75%, the highest so far reported for this type of reaction. Here the dominant contribution to stereoselectivity arises from the direct interaction between a donor group (the Dopa carboxylate) far from the substrate reaction site (the catechol ring) with the additional (third) copper center not involved in the oxidative catalysis. On the other hand, in the case of bulky substrates, such as L/D-catechin, the observed poor substrate recognition is associated with much weaker interactions between the chiral regions of the complex and the chiral part of the substrate.
*Recent biocatalytic oxidation–reduction cascades*
Schrittwieser, J.H.; Sattler, J.; Resch, V.; Mutti, F.G.; Kroutil, W.; Curr. Opin. Chem. Biol., 2011, 15, 249 – 256. DOI: 10.1016/j.cbpa.2010.11.010
moreThe combination of an oxidation and a reduction in a cascade allows performing transformations in a very economic and efficient fashion. The challenge is how to combine an oxidation with a reduction in one pot, either by running the two reactions simultaneously or in a stepwise fashion without isolation of intermediates. The broader availability of various redox enzymes nowadays has triggered the recent investigation of various oxidation–reduction cascades.
*Creating a biocatalyst for the production of an optically pure sterically hindered amine*
Mutti, F.G.; Sattler, J.; Tauber, K.; Kroutil, W.; ChemCatChem, 2011, 3, 109 - 111. DOI: 10.1002/cctc.201000349
more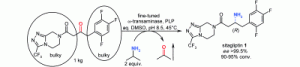
Amine for the top: A biocatalyst able to perform transamination of less hindered ketones is modified to transform a sterically demanding ketone substrate to the corresponding optically pure amine. This goal is achieved by rational analysis and design of the catalyst’s structure as well as other techniques of directed evolution. The biocatalyst is successfully adapted to the process and not vice versa.
*Ostensible enzyme promiscuity: Alkene cleavage by peroxidases*
Mutti, F.G.; Lara, M.; Kroutil, M.; Kroutil, W.; Chem. Eur. J., 2010, 16, 14142 - 14148. DOI: 10.1002/chem.201002265
more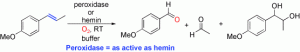
Enzyme promiscuity is generally accepted as the ability of an enzyme to catalyse alternate chemical reactions besides the ‘natural‘ one. In this paper peroxidases were shown to catalyse the cleavage of a C=C double bond adjacent to an aromatic moiety for selected substrates at the expense of molecular oxygen at an acidic pH. It was clearly shown that the reaction occurs due to the presence of the enzyme; furthermore, the reactivity was clearly linked to the hemin moiety of the peroxidase. Comparison of the transformations catalysed by peroxidase and by hemin chloride revealed that these two reactions proceed equally fast; additional experiments confirmed that the peptide backbone was not obligatory for the reaction and only a single functional group of the enzyme was required, namely in this case the prosthetic group (hemin). Consequently, we propose to define such a promiscuous activity as ‘ostensible enzyme promiscuity’. Thus, we call an activity that is catalysed by an enzyme ‘ostensible enzyme promiscuity’ if the reactivity can be tracked back to a single catalytic site, which on its own can already perform the reaction equally well in the absence of the peptide backbone.
*Simultaneous iridium catalysed oxidation and enzymatic reduction employing orthogonal reagents*
Mutti, F.G.; Orthaber, A.; Schrittwieser, J.H.; de Vries, J.G.; Pietschnig, R.; Kroutil, W.; Chem. Commun., 2010, 46, 8046 - 8048. DOI: 10.1039/c0cc02813d
more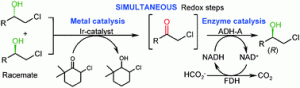
An iridium catalysed oxidation was coupled concurrently to an asymmetric biocatalytic reduction in one-pot; thus it was shown for the first time that iridium- and alcohol dehydrogenase-catalysed redox reactions are compatible. As a model system racemic chlorohydrins were transformed to enantioenriched chlorohydrins via an oxidation–asymmetric reduction sequence.
*Oxidative enzymatic alkene cleavage: Indications for a nonclassical enzyme mechanism*
Lara, M.; Mutti, F.G.; Glueck, S.M.; Kroutil, W.; J. Am. Chem. Soc., 2009, 131, 5368 - 5369. DOI: 10.1021/ja8097096
more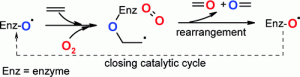
An enzyme preparation of Trametes hirsuta cleaves alkenes following neither the classical dioxygenase mechanism nor via a monooxygenase mechanism. A catalytic cycle for an alternative enzymatic alkene cleavage was proposed, whereby two oxygen atoms derived from two different oxygen molecules are incorporated into the product(s).
*Biomimetic modelling of copper enzymes: Synthesis, characterization, EPR analysis and enantioselective catalytic oxidations by a new chiral trinuclear copper(II) complex*
Mutti, F.G.; Zoppellaro, G.; Gullotti, M.; Santagostini, L.; Pagliarin, R.; Andersson, K.K.; Casella, L.; Eur. J. Inorg. Chem., 2009, 554 - 566. DOI: 10.1002/ejic.200800899
moreThe new octadentate ligand (R)-(+)-N,N′-dimethyl-N,N′-bis{-2-[bis(1-methyl-2-benzimidazolylmethyl)]-2-methylaminoethyl}-1,1′-binaphthyl-2,2′diamine [(R)-(+)-DABN-L-Ala-Bz4; L] was employed for the synthesis of dinuclear and trinuclear copper(II) complexes. The ligand design is based on the insertion of chiral residues derived from L-alanine between the diaminobinaphthyl (R)-(+)-DABN spacer and the aminobis(benzimidazole) metal binding units. The chiroptical properties of the ligand and the complexes are described. EPR experiments were performed on [Cu2L]4+ and [Cu3L]6+ at low temperatures. In the case of [Cu2L]4+, a weak dipolar interaction between the two spin centres was found. A similar weak spin interaction occurred in the trinuclear copper cluster, which could be treated likewise as a weakly coupled three-spin system. The analysis was substantiated by studying the complex EPR temperature behaviour, where population of the quartet state occurred only at high temperature (77 K), whereas at cryogenic temperatures (4 K) the system adopted a doublet state as a ground-state spin configuration. Titration with sodium azide of [Cu2L]4+ was consistent with terminal binding of one N3– molecule to each copper ion. Furthermore, dipolar interactions between spin centres were strongly suppressed in this case. For [Cu3L]6+, the adduct formed by the interaction with two azido molecules induced formation of a μ-azido bridges among the three Cu2+ ions and led to population of the quartet state even at cryogenic temperatures. This bridged configuration was, however, lost upon further addition of azido molecules. The copper(II) complexes were tested as catalysts in the oxidation of biogenic catechols and flavonoids by dioxygen to give the corresponding quinones, which were trapped as adducts with MBTH. The dinuclear complex [Cu2L]4+ displays poor substrate enantiodifferentiating ability, even though it exhibits catalytic activity comparable to that of [Cu3L]6+. The trinuclear complex [Cu3L]6+ exhibits significant enantioselectivity in the oxidations of the catecholamines L-/D-dopa methyl ester and L-/D-norepinephrine. The origin of this enantioselectivity must be associated with the mode of substrate binding, as it depends almost entirely on KM.
*Biomimetic modeling of copper complexes: A study of enantioselective catalytic oxidation on D-(+)-catechin and L-(−)-epicatechin with copper complexes*
Mutti, F.G.; Pievo, R.; Sgobba, M.; Gullotti, M.; Santagostini, L.; Bioinorganic chemistry and applications, 2008, Article ID 762029, 9 pages. DOI: 10.1155/2008/762029
moreThe biomimetic catalytic oxidations of the dinuclear and trinuclear copper(II) complexes versus two catechols, namely, D-(+)-catechin and L-(-)-epicatechin to give the corresponding quinones are reported. The unstable quinones were trapped by the nucleophilic reagent, 3-methyl-2-benzothiazolinone hydrazone (MBTH), and have been calculated the molar absorptivities of the different quinones. The catalytic efficiency is moderate, as inferred by kinetic constants, but the complexes exhibit significant enantio-differentiating ability towards the catechols, albeit for the dinuclear complexes, this enantio-differentiating ability is lower. In all cases, the preferred enantiomeric substrate is D-(+)-catechin to respect the other catechol, because of the spatial disposition of this substrate.
*Biocatalytic cleavage of alkenes with O2 and Trametes hirsuta G FCC 047*
Lara, M.; Mutti, F.G.; Glueck, S.M.; Kroutil, W.; Eur. J. Org. Chem., 2008, 3668 – 3672. DOI: 10.1002/ejoc.200800261
more
Alkenes possessing a C=C double bond adjacent to an aromatic ring were cleaved to yield the corresponding carbonyl compounds by use of molecular oxygen as the sole oxidant and a cell-free extract of the wood-degrading fungus Trametes hirsuta FCC 047 as catalyst. The oxygen pressure required was optimized. Special adapted equipment allowed 96 reactions to be performed in parallel under controlled oxygen pressure. A broad spectrum of aryl-alkenes was successfully converted into the corresponding ketones/aldehydes with excellent chemoselectivity under a controlled oxygen atmosphere (2 bar).