


January 2018-1: Towards new green chemistry using enzymes and light
Combining photocatalysis and biocatalysis for selective functionalization of carbon-hydrogen bonds
Chemists at the Biocatalysis group of the University of Amsterdam’s Van ‘t Hoff Institute for Molecular Sciences (HIMS) have contributed to research that potentially renders the synthesis of certain pharmaceuticals, cosmetics and agrochemicals much simpler, cheaper and more sustainable. The results of this international project, combining photocatalysts and natural enzymes, have just been published by Nature Catalysis.
Photo-biocatalysis is a novel concept that aims at merging the best features from photocatalysis and enzymatic catalysis. In the research published in Nature Catalysis scientists have exploited visible light as source of energy to generate small but reactive H2O2 molecules, which then serve as co-substrates for a series of complex enzymatic transformations.
The project was a collaboration between several groups in the Netherlands (TU Delft, UvA), Spain (CSIC, Madrid) and Germany (Forschungszentrum Jülich) and focused on the selective functionalization of carbon-hydrogen bonds, which is a long sought-after chemical transformation with great industrial relevance. The UvA, HIMS Biocatalysis group contributed with their expertise in enzymatic amination and halogenation.
Selective transformations
Starting point for the carbon-hydrogen functionalization is the insertion of an oxygen atom into the carbon-hydrogen bond, creating an alcohol (hydroxyl) group that can be functionalized further. In traditional synthetic chemistry, the high reactivity of oxygen-transfer reagents is the main challenge here. When using these reagents, the chemo- and regioselectivity of the reaction is very difficult to control. Enzymes provide a great advantage as they are well-known for their ability to carry out chemical transformations with exquisite selectivities.
The enzymatic hydroxylation of isolated carbon-hydrogen bonds has already been applied in industry using monooxygenases (P450). However, due to its complex working mechanism, P450 is not very widely applicable. The biocatalytic alternative to P450 monooxygenases is the use of peroxygenases. These are capable of selective insertion of an oxygen atom into carbon-hydrogen bonds at the expense of hydrogen peroxide (H2O2). The problem here is, nonetheless, that the practical applicability of peroxygenases requires the in situ generation of H2O2. That way a low and constant H2O2 concentration is maintained during the course of the reaction, hence preventing loss of enzyme activity. The in situ generation of H2O2 can be achieved through enzymatic oxidation of glucose catalyzed by glucose oxidase, but this is a quite inefficient process: for each generated molecule of H2O2 a glucose molecule is wasted. Adding to this, using glucose is not desirable from a sustainability perspective.
Water splitting
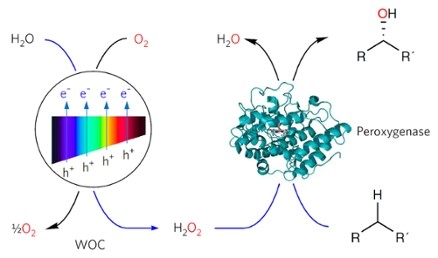
Fig. 1. A water oxidation catalyst (WOC) mediates the photochemical oxidation of water to produce H2O2, which is utilized by a peroxygenase to catalyse (stereo)selective oxyfunctionalization reactions. Image from: Nature Catalysis.
A photo-biocatalytic process was devised, where the H2O2 co-substrate is efficiently produced in situ from water and light using titanium oxide water oxidation catalysts (WOC’s). These are widely researched for their ability to split water and generate hydrogen (H2). Under aerobic conditions these WOC’s can also form H2O2. This feature was exploited to design a system for the light-driven enzymatic selective functionalization of carbon-hydrogen bonds.
In this system, the WOC’s provide H2O2 to a robust peroxygenase that was recombinantly produced from the fungus Agrocybe aegerita. The system achieves appealing results with the selective hydroxylation of a broad range of aliphatic and aromatic alkanes. For instance, the epoxidation of ethyl benzene to (R)-1-phenyl ethanol yielded the alcohol product in >98% enantiomeric excess with turnover numbers of 16000 – 21000.
Second generation process
A second generation was then developed. It turned out that by tuning of the reaction conditions the original system could be triggered to a formal ‘over-oxidation’ of the carbon-hydrogen bond so that ketones were produced rather than alcohols. These were subsequently converted to amines in a one-pot process using the stereocomplementary aminotransferase enzymes. This permitted access to both (R) and (S)-configured amines in enantiopure form.
Fig. 2. Schematic representation of the second generation process. The water splitting light-driven TiO2 catalyst (centre) produces H2O2 to ‘fuel’ the enzymatic conversion of C-H bonds to alcohols (left). The latter are further oxidized to yield ketones, which are subsequently enzymatically aminated (right).
The study was further complemented by using vanadium-peroxidase enzymes that catalyze the halogenation of aromatic compounds. The in situ H2O2 generation system was applied to the V-dependent haloperoxidase from Curvularia inaequalis and thymol as test substrate. Selective bromination was successfully conducted to afford the para-bromo substituted isomer in ca. 63% conversion.
More complex systems
The researchers expect that further optimization of reaction conditions, the study of more bio-compatible water oxidation catalysts, and the stabilization of enzymes (by protein engineering and immobilization techniques) can open the way to the application of the photo-biocatalytic cascades. The current work may also serve as model for the creation of more complex photo-bio hybrid systems for even more challenging chemical transformations.
Article
Wuyuan Zhang, Elena Fernández-Fueyo, Yan Ni, Morten van Schie, Jenö Gacs, Rokus Renirie, Ron Wever, Francesco G. Mutti, Dörte Rother, Miguel Alcalde & Frank Hollmann: Selective aerobic oxidation reactions using a combination of photocatalytic water oxidation and enzymatic oxyfunctionalizations. Nature Catalysis (2017), Published online 20 November 2017. DOI: 10.1038/s41929-017-0001-5